Generalidad
El ADN mitocondrial, o ADNmt, es el ácido desoxirribonucleico que reside dentro de las mitocondrias, es decir, los orgánulos de las células eucariotas responsables del muy importante proceso celular de fosforilación oxidativa.

Sin embargo, también tiene algunas peculiaridades, tanto estructurales como funcionales, que la hacen única en su género. Estas peculiaridades incluyen: la circularidad de la doble hebra de nucleótidos, el contenido de genes (que es solo de 37 elementos) y la ausencia casi total de secuencias de nucleótidos no codificantes.
El ADN mitocondrial cumple una función fundamental para la supervivencia de las células: produce las enzimas necesarias para la realización de la fosforilación oxidativa.
¿Qué es el ADN mitocondrial?
El ADN mitocondrial, o ADNmt, es el ADN ubicado dentro de las mitocondrias.
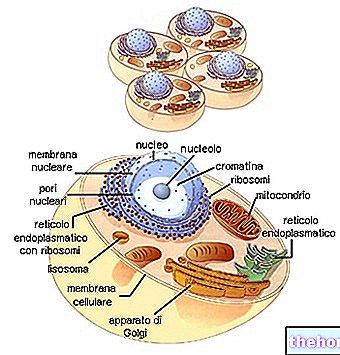
Las mitocondrias son esos grandes orgánulos celulares, típicos de los organismos eucariotas, que convierten la energía química contenida en los alimentos en ATP, que es una forma de energía que pueden aprovechar las células.
ANTECEDENTES DE LA ESTRUCTURA Y FUNCIONAMIENTO DE LAS MITOCONDRONES
De forma tubular, filamentosa o granular, las mitocondrias residen en el citoplasma, ocupando casi el 25% del volumen de este último.
Tienen dos membranas bicapa de fosfolípidos, una más externa y otra más interna.
La membrana más externa, conocida como membrana mitocondrial externa, representa el perímetro de cada mitocondria y tiene proteínas de transporte (porinas y más), que la hacen permeable a moléculas de un tamaño igual o menor a 5,000 daltons.
La membrana más interna, conocida como membrana mitocondrial interna, contiene todos los componentes enzimáticos (o enzimáticos) y coenzimáticos, necesarios para la síntesis de ATP, y define un espacio central, llamado matriz.
A diferencia de la membrana más externa, la membrana mitocondrial interna tiene numerosas invaginaciones, las llamadas crestas, que aumentan su área total.
Entre las dos membranas mitocondriales hay un espacio de cerca de 60-80 Angstroms (A), este espacio se llama espacio intermembrana El espacio intermembrana tiene una composición muy similar a la del citoplasma.
La síntesis de ATP, operada por las mitocondrias, es un proceso muy complejo, que los biólogos identifican con el término fosforilación oxidativa.
UBICACIÓN PRECISA DEL ADN MITOCONDRAL Y CANTIDAD

Figura: una mitocondria humana.
El ADN mitocondrial reside en la matriz mitocondrial, es decir, en el espacio delimitado por la membrana mitocondrial interna.
Según estudios científicos fiables, cada mitocondria puede contener de 2 a 12 copias de ADN mitocondrial.
Dado que, en el cuerpo humano, algunas células pueden contener varios miles de mitocondrias dentro de ellas, el número total de copias de ADN mitocondrial en una sola célula humana puede alcanzar hasta 20.000 unidades.
tenga en cuenta: el número de mitocondrias en las células humanas varía según el tipo de célula. Por ejemplo, los hepatocitos (es decir, células del hígado) pueden contener entre 1000 y 2000 mitocondrias cada uno, mientras que los eritrocitos (es decir, glóbulos rojos) están totalmente desprovistos de ellos.
Estructura
La estructura general de una molécula de ADN mitocondrial se asemeja a la estructura general del ADN nuclear, es decir, la herencia genética presente dentro del núcleo de las células eucariotas.
En efecto, análogamente al ADN nuclear:
- El ADN mitocondrial es un biopolímero que consta de dos cadenas largas de nucleótidos. Los nucleótidos son moléculas orgánicas, resultado de la unión de tres elementos: un azúcar con 5 átomos de carbono (en el caso del ADN, desoxirribosa), una base nitrogenada y un grupo fosfato.
- Cada nucleótido del ADN mitocondrial se une al siguiente nucleótido de la misma hebra por medio de un enlace fosfodiéster entre el carbono 3 de su desoxirribosa y el grupo fosfato del nucleótido inmediatamente siguiente.
- Las dos hebras de ADN mitocondrial tienen orientaciones opuestas, con el extremo de una interactuando con el extremo de la otra y viceversa. Esta disposición particular se conoce como disposición antiparalela (u orientación antiparalela).
- Las dos cadenas de ADN mitocondrial interactúan entre sí a través de las bases nitrogenadas.
Específicamente, cada base nitrogenada de cada filamento establece enlaces de hidrógeno con una y solo una base nitrogenada, presente en el otro filamento.
Este tipo de interacción se denomina "emparejamiento entre bases nitrogenadas" o "par de bases nitrogenadas". - Las bases nitrogenadas del ADN mitocondrial son adenina, timina, citosina y guanina.
El emparejamiento al que dan lugar estas bases nitrogenadas no es aleatorio, sino muy específico: la adenina interactúa solo con la timina, mientras que la citosina solo interactúa con la guanina. - El ADN mitocondrial alberga genes (o secuencias de genes). Los genes son secuencias de nucleótidos más o menos largos, con un significado biológico bien definido. En la mayoría de los casos dan lugar a proteínas.

PARTICULARIDADES ESTRUCTURALES DEL ADN MITOCONDRAL
Más allá de las analogías antes mencionadas, el ADN mitocondrial humano tiene algunas peculiaridades estructurales, que lo distinguen considerablemente del ADN nuclear humano.
Primero, es una molécula circular, mientras que el ADN nuclear es una molécula lineal.
Por lo tanto, tiene 16.569 pares de bases nitrogenadas, mientras que el ADN nuclear tiene la friolera de 3.300 millones.
Contiene 37 genes, mientras que el ADN nuclear parece contener entre 20.000 y 25.000.
No está organizado en cromosomas, mientras que el ADN nuclear se divide en 23 cromosomas y forma, con algunas proteínas específicas, una sustancia llamada cromatina.
Finalmente, incluye una serie de nucleótidos que participan en dos genes al mismo tiempo, mientras que el ADN nuclear tiene genes cuyas secuencias de nucleótidos están bien definidas y son distintas entre sí.
Origen
Lo más probable es que el ADN mitocondrial tenga un origen "bacteriano".
De hecho, sobre la base de numerosos estudios independientes, los biólogos moleculares creen que la presencia celular de ADN mitocondrial es el resultado de la incorporación, por parte de células eucariotas ancestrales, de organismos bacterianos independientes, muy similares a las mitocondrias.
Este curioso descubrimiento ha sorprendido solo parcialmente a la comunidad científica, ya que el ADN presente en las bacterias es generalmente una cadena de nucleótidos circular, como el ADN mitocondrial.
La teoría según la cual las mitocondrias y el ADN mitocondrial tienen un "origen bacteriano" toma el nombre de "teoría endosimbiótica", de la palabra "endosimbiosis". Brevemente, en biología, el término "endosimbiosis" indica una colaboración entre dos organismos, que implica la "incorporación de uno en el otro, con el fin de obtener una cierta ventaja.
Curiosidad
Según estudios científicos fiables, en el curso de la evolución muchos genes bacterianos, presentes en el futuro ADN mitocondrial, habrían cambiado de ubicación, pasando al ADN nuclear.
En otras palabras, al comienzo de la endosimbiosis, algunos genes ahora presentes en el ADN nuclear residían en el ADN de esos organismos bacterianos, que luego se convertirían en mitocondrias.
Para apoyar la teoría relacionada con un cambio de genes entre el ADN mitocondrial y el ADN nuclear, está la observación de que ciertos genes se derivan del ADN mitocondrial, en algunas especies, y del ADN nuclear, en otras.
Función
El ADN mitocondrial produce enzimas (es decir, proteínas) necesarias para la correcta implementación del delicado proceso de fosforilación oxidativa.
Las instrucciones para sintetizar estas enzimas residen en los 37 genes que componen el genoma del ADN mitocondrial.
QUÉ CÓDIGO DE LOS GENES DEL ADN MITOCONDRAL: LOS DETALLES
Los 37 genes del ADN mitocondrial codifican: proteínas, ARNt y ARNr.
En particular:
- 13 codifican 13 proteínas encargadas de llevar a cabo la fosforilación oxidativa
- 22 código para 22 moléculas de ARNt
- 2 codifican 2 moléculas de ARNr
Las moléculas de tRNA y rRNA son fundamentales para la síntesis de las 13 proteínas mencionadas, ya que forman la maquinaria que regula su producción.
Entonces, en otras palabras, el ADN mitocondrial posee la información para producir un cierto conjunto de proteínas y las herramientas necesarias para su síntesis.
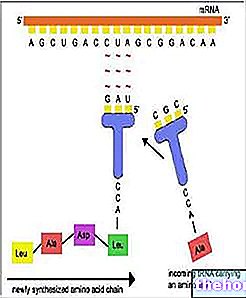
¿Qué son el ARN, el ARNt y el ARNr??
El ARN, o ácido ribonucleico, es el ácido nucleico que juega un papel fundamental en la generación de proteínas, a partir del ADN.
Generalmente monocatenario, el ANN puede existir en varias formas (o tipos), dependiendo de la función específica a la que se delega.
TRNA y rRNA son dos de estas posibles formas.
El ARNt se utiliza para agregar aminoácidos durante el proceso de fabricación de proteínas Los aminoácidos son las unidades moleculares que componen las proteínas.
El ARNr forma los ribosomas, es decir, las estructuras celulares en las que tiene lugar la síntesis de proteínas.
Para conocer en detalle la ANN y sus funciones, los lectores pueden pinchar aquí.
DETALLES FUNCIONALES DEL ADN MITOCONDRAL
Desde un punto de vista funcional, el ADN mitocondrial tiene algunas características peculiares que lo distinguen claramente del ADN nuclear.
Aquí es en lo que consisten estas características peculiares:
- El ADN mitocondrial es semiindependiente, en el sentido de que necesita la intervención de algunas proteínas sintetizadas a partir del ADN nuclear.
Por otro lado, el ADN nuclear es completamente autónomo y produce por sí mismo todo lo que necesita para realizar adecuadamente sus tareas. - El ADN mitocondrial tiene un código genético ligeramente diferente al del ADN nuclear. Esto conduce a una serie de diferencias en la producción de proteínas: si una determinada secuencia de nucleótidos en el ADN nuclear conduce a la creación de una determinada proteína, la misma secuencia en el ADN mitocondrial conduce a la formación de una proteína ligeramente diferente.
- El ADN mitocondrial tiene muy pocas secuencias de nucleótidos no codificantes, es decir, no producen proteínas, ARNt o ARNr. En términos porcentuales, solo el 3% del ADN mitocondrial no es codificante.
Por otro lado, el ADN nuclear tiene solo un 7% de codificación, por lo que contiene muchas secuencias de nucleótidos no codificantes (hasta un 93%).
Tabla: resumen de las diferencias entre el ADN mitocondrial humano y el ADN nuclear humano.
ADN mitocondrial
ADN nuclear
- Es circular
- Es lineal
- Tiene 16,569 pares de bases nitrogenadas en total
- Tiene un total de 3.300 millones de pares de bases nitrogenadas.
- Contiene 37 genes en total
- Contiene entre 20.000 y 25.000 genes.
- Para funcionar correctamente, necesita el apoyo de algunos productos genéticos, derivados del ADN nuclear.
- Es autónomo y produce por sí mismo todo lo que necesita para realizar adecuadamente sus funciones.
- Puede estar presente en varias copias dentro de cada mitocondria individual.
- Es único, es decir, está en una sola copia y reside en el núcleo
- El 97% de la secuencia de nucleótidos que lo componen está codificando
- Solo el 7% de la secuencia de nucleótidos que lo componen está codificando
- No está organizado en cromosomas.
- Está dividido en 23 cromosomas.
- Utiliza un código genético ligeramente diferente del, por así decirlo, "tradicional".
- Utilice el código genético "tradicional"
- Su herencia es materna
- Su herencia es mitad materna y mitad paterna.
- Algunos de sus nucleótidos participan en dos genes al mismo tiempo.
- Las secuencias de nucleótidos que componen los genes se distinguen bien entre sí.
Herencia
La herencia del ADN mitocondrial es estrictamente materna.
Esto significa que, en un par de padres, es la mujer quien transmite el ADN mitocondrial a la progenie (es decir, a los hijos).
De manera completamente opuesta a lo anterior, la herencia del ADN nuclear es mitad materna y mitad paterna, es decir, ambos padres contribuyen por igual a la transmisión del ADN nuclear en la descendencia.
tenga en cuenta: la herencia materna del ADN mitocondrial también involucra la estructura mitocondrial. Por tanto, las mitocondrias presentes en un individuo son maternas.
Patologías asociadas
Premisa: Una mutación genética es un cambio permanente en la secuencia de nucleótidos, que forman un gen de ADN nuclear o mitocondrial.
Normalmente, la presencia de una mutación genética da como resultado una "alteración o pérdida de la función normal del gen implicado".
La presencia de mutaciones en los genes del ADN mitocondrial puede provocar una amplia gama de enfermedades, que incluyen:
- Neuropatía óptica hereditaria de Leber
- Síndrome de Kearns-Sayre
- Síndrome de Leigh
- La deficiencia de citocromo C oxidasa
- Oftalmoplejía externa progresiva
- Síndrome de Pearson
- Encefalomiopatía mitocondrial con acidosis láctica y episodios similares a accidentes cerebrovasculares (síndrome MELAS)
- Diabetes con sordera de transmisión materna
- Epilepsia mioclónica con fibras rojas irregulares.
En cuanto a las condiciones patológicas vinculadas a una o más mutaciones del ADN mitocondrial, es necesario aclarar dos aspectos.
En primer lugar, la gravedad de la enfermedad depende de la relación cuantitativa entre el ADN mitocondrial mutado y el ADN mitocondrial normal y sano. Si el número de ADN mitocondrial mutado es mucho mayor que el de ADN sano, la condición resultante será más grave.
En segundo lugar, las mutaciones en el ADN mitocondrial solo afectan a algunos tejidos del organismo, en particular a aquellos que requieren grandes cantidades de ATP producto del proceso de fosforilación oxidativa. Esto es bastante comprensible: sufrir más de un mal funcionamiento del ADN mitocondrial son las células que más necesitan la función que normalmente cumple el ADN mitocondrial.
NEUROPATÍA ÓPTICA HEREDITARIA DE LEBER
La neuropatía óptica hereditaria de Leber surge como resultado de la mutación de hasta cuatro genes del ADN mitocondrial. Estos genes contienen la información que conduce a la síntesis del llamado complejo I (o NADH óxido-reductasa), una de las diversas enzimas implicadas en el proceso de fosforilación oxidativa.
Las manifestaciones de la patología consisten en una degeneración progresiva del nervio óptico y una pérdida gradual de la visión.
SÍNDROME DE KEARNS-SAYRE
El síndrome de Kearns-Sayre aparece debido a la falta de una cantidad considerable de ADN mitocondrial (Nota: la falta de una determinada secuencia de nucleótidos se llama deleción).
Las personas con síndrome de Kearns-Sayre desarrollan oftalmoplejía (parálisis total o parcial de los músculos oculomotores), una forma de retinopatía y anomalías del ritmo cardíaco (bloqueo auriculoventricular).
SÍNDROME DE LEIGH
El síndrome de Leigh surge como resultado de mutaciones en el ADN mitocondrial, que pueden afectar la proteína ATP-sintasa (también llamada complejo V) y / o algunos ARNt.
El síndrome de Leigh es una enfermedad neurológica progresiva, que aparece en la infancia o la niñez y es responsable de: retraso en el desarrollo, debilidad muscular, neuropatía periférica, trastornos motores, dificultades respiratorias y oftalmoplejía.
DEFICIENCIA DE CITOCROMO C OXIDASA
La deficiencia de citocromo C oxidasa ocurre debido a la mutación de al menos 3 genes del ADN mitocondrial. Estos genes son esenciales para la correcta síntesis de la enzima citocromo C oxidasa (o complejo IV), involucrada en el proceso de fosforilación oxidativa.
Las manifestaciones típicas de la deficiencia de citocromo C oxidasa consisten en: disfunción del músculo esquelético, disfunción cardíaca, disfunción renal y disfunción hepática.
OFTALMOPLEGIA EXTERNA PROGRESIVA
La oftalmoplejía externa progresiva surge de la falta de una cantidad sustancial de nucleótidos del ADN mitocondrial (deleción)
De carácter progresivo (como se desprende del nombre), esta patología provoca una parálisis de los músculos oculomotores, con la consecuente ptosis y problemas visuales considerables.
SÍNDROME DE PEARSON
El síndrome de Pearson aparece después de una deleción notoria del ADN mitocondrial, de manera similar a la oftalmoplejía externa progresiva y el síndrome de Kearns-Sayre.
Las manifestaciones típicas del síndrome de Pearson consisten en: anemia sideroblástica, disfunción pancreática (por ejemplo, diabetes insulinodependiente), déficits neurológicos y trastornos musculares.
El síndrome de Pearson generalmente hace que la persona afectada muera a una edad temprana. De hecho, los afectados por esta patología rara vez llegan a la edad adulta.
SÍNDROME DE MELAS
El síndrome de MELAS, también conocido como encefalomiopatía mitocondrial con acidosis láctica y episodios similares a un accidente cerebrovascular, surge de la mutación de al menos 5 genes del ADN mitocondrial.
Estos genes contribuyen a la síntesis de NADH óxido-reductasa, o complejo I, y de algunos ARNt.
El síndrome de MELAS implica la presencia de trastornos neurológicos, trastornos musculares, acumulación inusual de ácido láctico en los tejidos (con todos los síntomas que lo acompañan), problemas respiratorios, pérdida del control de la función intestinal, fatiga recurrente, problemas renales, problemas cardíacos, diabetes, epilepsia y falta de cordinacion.
OTRAS PATOLOGÍAS
Según diversos estudios científicos, enfermedades como el síndrome de vómitos cíclicos, la retinitis pigmentosa, la ataxia, la enfermedad de Parkinson y la enfermedad de Alzheimer también verían la participación del ADN mitocondrial y algunas de sus mutaciones.