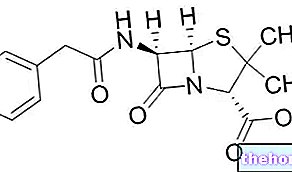
Ingredientes activos: Temozolomida
Temodal 5 mg cápsulas duras
Temodal 20 mg cápsulas duras
Temodal 100 mg cápsulas duras
Temodal 140 mg cápsulas duras
Temodal 180 mg cápsulas duras
Temodal 250 mg cápsulas duras
¿Por qué se usa Temodal? ¿Para qué sirve?
Temodal contiene un medicamento llamado temozolomida. Este medicamento es un agente contra el cáncer.
Temodal se usa para tratar formas específicas de tumor cerebral:
- en adultos con glioblastoma multiforme diagnosticado por primera vez. Temodal se utiliza inicialmente en combinación con radioterapia (fase de tratamiento concomitante) y posteriormente solo (fase de tratamiento en monoterapia).
- en niños de 3 años o más y en pacientes adultos con glioma maligno, como glioblastoma multiforme o astrocitoma anaplásico. Temodal se usa en estos cánceres que recaen o progresan después de la terapia estándar.
Contraindicaciones Cuándo no se debe usar Temodal
No tome Temodal
- si es alérgico a la temozolomida oa cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- si ha tenido una reacción alérgica previa a la dacarbazina (un medicamento contra el cáncer, a veces llamado DTIC). Los signos de una reacción alérgica incluyen picazón, dificultad para respirar o sibilancias, hinchazón de la cara, labios, lengua o garganta.
- si el número de ciertos tipos de células sanguíneas está muy reducido (mielosupresión), como el número de glóbulos blancos y plaquetas. Estas células sanguíneas son importantes para combatir infecciones y para una adecuada coagulación de la sangre. Su médico le hará análisis de sangre para asegurarse de que haya suficientes células para iniciar el tratamiento.
Precauciones de uso Qué necesita saber antes de tomar Temodal
Consulte a su médico, farmacéutico o enfermero antes de tomar Temodal.
- ya que debe observarse cuidadosamente para detectar el desarrollo de una forma grave de infección del pecho llamada neumonía por Pneumocystis jirovecii (PCP). Si es un paciente diagnosticado por primera vez (glioblastoma multiforme), se le puede administrar Temodal durante 42 días en combinación con radioterapia. En este caso, su médico también le recetará medicamentos para ayudarlo a prevenir este tipo de neumonía (PCP).
- si alguna vez ha tenido o puede tener actualmente una infección por hepatitis B. Esto se debe a que Temodal puede hacer que la hepatitis B se reactive, lo que en algunos casos puede ser fatal. Antes de iniciar el tratamiento, su médico examinará cuidadosamente a los pacientes para comprobar si existen signos de esta infección.
- si tiene un número bajo de glóbulos rojos (anemia), glóbulos blancos y plaquetas, o problemas de coagulación de la sangre antes de comenzar el tratamiento, o si los desarrolla durante el tratamiento. Su médico puede decidir reducir su dosis, suspender, suspender o cambiar su tratamiento.También puede necesitar otros tratamientos.En algunos casos, puede ser necesario dejar de tomar Temodal.Se analizarán muestras de sangre con frecuencia durante el tratamiento para comprobar los efectos secundarios de Temodal en sus células sanguíneas.
- ya que puede tener un riesgo bajo de desarrollar otros trastornos de las células sanguíneas, incluida la leucemia.
- Si tiene náuseas (malestar estomacal) y / o vómitos, que son efectos adversos muy frecuentes de Temodal (ver sección 4), su médico puede recetarle un medicamento (un antiemético) para ayudar a prevenir los vómitos. Si vomita con frecuencia antes o durante el tratamiento, pregunte a su médico cuál es el mejor momento para tomar Temodal hasta que los vómitos estén bajo control. Si vomita después de tomar una dosis, no tome una segunda el mismo día.
- si tiene fiebre o síntomas de una infección, póngase en contacto con su médico inmediatamente.
- Si tiene más de 70 años, puede ser más propenso a sufrir infecciones, hematomas o hemorragias.
- Si tiene problemas de hígado o riñón, es posible que deba ajustar su dosis de Temodal.
Niños y adolescentes
No administre este medicamento a niños menores de 3 años ya que no se ha estudiado. Existe información limitada en pacientes mayores de 3 años que han tomado Temodal.
Interacciones ¿Qué medicamentos o alimentos pueden cambiar el efecto de Temodal?
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
Advertencias Es importante saber que:
Embarazo, lactancia y fertilidad
Si está embarazada, cree que podría estar embarazada o tiene intención de quedarse embarazada, consulte a su médico o farmacéutico antes de utilizar este medicamento. Esto se debe a que no debe tratarse con Temodal durante el embarazo a menos que su médico lo indique claramente.
Tanto los pacientes masculinos como femeninos que estén tomando Temodal deben tomar precauciones anticonceptivas eficaces (ver también "Fertilidad masculina" a continuación).
Debe interrumpir la lactancia si está en tratamiento con Temodal.
Fertilidad masculina
Temodal puede causar infertilidad permanente. Los pacientes varones deben utilizar métodos anticonceptivos eficaces y no intentar tener un hijo durante los 6 meses posteriores a la interrupción del tratamiento. Se recomienda consultar sobre el almacenamiento de esperma antes de iniciar el tratamiento.
Conducción y uso de máquinas
Temodal puede hacer que se sienta cansado o con sueño. En este caso, no conduzca ni utilice herramientas, máquinas o bicicletas hasta que vea los efectos que este medicamento tiene en usted (ver sección 4).
Temodal contiene lactosa
Temodal contiene lactosa (un tipo de azúcar). Si su médico le ha dicho que padece una "intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento".
Posología y forma de empleo Cómo utilizar Temodal: Posología
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte a su médico o farmacéutico.
Posología y duración del tratamiento.
Su médico determinará su dosis de Temodal. Depende de su tamaño (altura y peso) y, si tiene un tumor recurrente, de cualquier tratamiento de quimioterapia anterior.
Se pueden recetar otros medicamentos (antieméticos) antes y / o después de tomar Temodal para prevenir o controlar las náuseas y los vómitos.
Pacientes con glioblastoma multiforme diagnosticados por primera vez:
Si es un paciente al que se le ha diagnosticado cáncer por primera vez, el tratamiento se realizará en dos etapas:
- Temodal se asocia inicialmente con radioterapia (fase concomitante)
- A partir de entonces, Temodal se administra solo (fase de monoterapia).
Durante la fase concomitante, su médico iniciará Temodal a una dosis de 75 mg / m2 (dosis habitual). Tomará esta dosis todos los días durante 42 días (hasta un máximo de 49 días) en combinación con radioterapia. Su dosis de Temodal puede retrasarse o interrumpirse en función del número de células sanguíneas y de cómo esté manejando el medicamento durante la fase concomitante.
Una vez que se complete la radioterapia, suspenderá el tratamiento durante 4 semanas. Esto le dará a su cuerpo la oportunidad de recuperarse.
Luego, comenzará la monoterapia.
Durante la fase de monoterapia, la dosis y la forma de tomar Temodal serán diferentes. Su médico calculará la dosis correcta. Puede haber hasta 6 períodos de tratamiento (ciclos). Cada uno dura 28 días. Tomará su nueva dosis de Temodal solo una vez al día durante los primeros 5 días ("días de terapia") de cada ciclo. La primera dosis será de 150 mg / m 2. Luego seguirán 23 días sin Temodal. Así se alcanzan los 28 días del ciclo de tratamiento.
Después del día 28, comenzará el siguiente ciclo. Volverá a tomar Temodal una vez al día durante 5 días seguido de 23 días sin Temodal. La dosis de Temodal puede ajustarse, retrasarse o interrumpirse en función de su recuento sanguíneo y de cómo maneja el medicamento durante cada ciclo de tratamiento.
Pacientes con tumores que han vuelto o han empeorado (gliomas malignos, como glioblastoma multiforme o astrocitoma anaplásico) tratados con Temodal solo:
Un curso de tratamiento con Temodal dura 28 días. Tomará Temodal solo una vez al día durante los primeros 5 días. Esta dosis diaria depende de cualquier tratamiento de quimioterapia previo.
Si no ha sido tratado previamente con quimioterapia, su primera dosis de Temodal será de 200 mg / m2 una vez al día durante los primeros 5 días. Si ha sido tratado previamente con quimioterapia, su primera dosis de Temodal será de 150 mg / m 2 una vez al día durante los primeros 5 días.
Durante los próximos 23 días, dejará de recibir Temodal. Esto completará el ciclo de 28 días.
Después del día 28, comenzará el siguiente ciclo. Volverá a tomar Temodal una vez al día durante cinco días, seguido de 23 días sin Temodal.
Antes de cada nuevo ciclo, se le realizarán análisis de sangre para ver si es necesario cambiar la dosis de Temodal. Según los resultados de sus análisis de sangre, su médico puede cambiar su dosis para el próximo ciclo.
Cómo tomar Temodal
Tome la dosis prescrita de Temodal una vez al día, preferiblemente a la misma hora todos los días.
Tome las cápsulas con el estómago vacío; por ejemplo, al menos una hora antes del desayuno.
Trague las cápsulas enteras con un vaso de agua. No abra, triture ni mastique las cápsulas. Si la cápsula está dañada, evite el contacto del polvo con la piel, los ojos o la nariz. ojos o nariz, enjuague el área afectada con agua.
Dependiendo de la dosis prescrita, puede ser necesario tomar más de una cápsula al mismo tiempo, posiblemente con diferentes concentraciones (contenido del principio activo en mg). El color de la cubierta de la cápsula es diferente para cada concentración (consulte la tabla a continuación).
Debe asegurarse de comprender y recordar lo siguiente:
- cuántas cápsulas necesita tomar para su dosis diaria. Pídale a su médico o farmacéutico que lo anote (incluido el color).
- cuales son los dias de tomar la terapia.
Revise la dosis con su médico cada vez que tenga que comenzar un nuevo ciclo, ya que puede ser diferente del ciclo anterior.
Siga exactamente las instrucciones de administración de Temodal indicadas por su médico. En caso de duda, consulte a su médico o farmacéutico. Los errores al tomar este medicamento pueden causar daños graves a su salud.
Si olvidó tomar Temodal
Tome la dosis omitida lo antes posible el mismo día. Si ha pasado un día completo, comuníquese con su médico. No tome una dosis doble para compensar las dosis olvidadas, a menos que su médico se lo indique.
Si tiene cualquier otra duda sobre el uso de este medicamento, consulte a su médico, farmacéutico o enfermero.
Sobredosis Qué hacer si ha tomado demasiado Temodal
Si accidentalmente toma más cápsulas de las recetadas, comuníquese con su médico, farmacéutico o enfermero inmediatamente.
Efectos secundarios ¿Cuáles son los efectos secundarios de Temodal?
Como todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Comuníquese con su médico de inmediato si experimenta:
- una reacción alérgica grave (hipersensibilidad) (urticaria, sibilancias u otra dificultad para respirar),
- sangrado incontrolado,
- ataques (convulsiones),
- fiebre,
- dolor de cabeza severo que no desaparece.
El tratamiento con Temodal puede provocar una reducción de ciertos tipos de células sanguíneas. Esto puede provocar un aumento de hematomas o sangrado, anemia (disminución del número de glóbulos rojos), fiebre y reducción de la resistencia a las infecciones. La reducción del número de células sanguíneas suele ser de corta duración. En algunos casos, puede prolongarse y provocar una forma muy grave de anemia (anemia aplásica). Su médico controlará su sangre con regularidad y decidirá si es necesaria una terapia específica. En algunos casos, se reducirá la dosis de Temodal o se terminará el tratamiento.
Efectos secundarios de los estudios clínicos:
Temodal en tratamiento combinado con radioterapia en glioblastoma de nuevo diagnóstico
Los pacientes que reciben Temodal en combinación con radioterapia pueden experimentar efectos secundarios diferentes a los notificados por los pacientes que reciben Temodal solo. Los siguientes efectos secundarios pueden ocurrir y pueden requerir atención médica.
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
pérdida de apetito, dolor de cabeza, estreñimiento (dificultad para defecar), náuseas (malestar del estómago), vómitos, erupción cutánea, caída del cabello, cansancio.
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
infecciones de la boca, infecciones de heridas, recuento reducido de células sanguíneas (neutropenia, trombocitopenia, linfopenia, leucopenia), aumento de azúcar en sangre, pérdida de peso, cambios en el estado mental o el estado de alerta, ansiedad / depresión, somnolencia, dificultad para hablar, problemas de equilibrio, mareos, confusión , pérdida de memoria, dificultad para concentrarse, incapacidad para conciliar el sueño o permanecer dormido, sensación de hormigueo, hematomas, temblores, visión anormal o confusa, visión doble, problemas de audición, dificultad para respirar, tos, coágulos de sangre en las piernas, retención de líquidos, piernas hinchadas , diarrea, dolor de estómago o abdominal, ardor de estómago, malestar estomacal, dificultad para tragar, boca seca, irritación o enrojecimiento de la piel, piel seca, picazón, debilidad dolor muscular, dolor en las articulaciones, dolores y molestias musculares, orinar con frecuencia, dificultad para retener la orina, reacción alérgica , fiebre, lesión de rueda de radio pia, hinchazón de la cara, dolor, alteración del gusto, pruebas de función hepática anormales.
Poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
síntomas de la gripe, manchas rojas subcutáneas, bajo nivel de potasio en sangre, aumento de peso, cambios de humor, alucinaciones y alteraciones de la memoria, parálisis parcial, alteraciones de la coordinación, alteraciones sensoriales, pérdida parcial de la visión, ojos secos o dolorosos, sordera, infección del oído medio, zumbido en el oídos, dolor de oído, palpitaciones (cuando puede oír los latidos del corazón), coágulos de sangre en el pulmón, presión arterial alta, neumonía, inflamación de las fosas nasales, bronquitis, resfriado o gripe, distensión del estómago, dificultad para controlar las deposiciones, hemorroides, descamación de la piel. , aumento de la sensibilidad de la piel a la luz solar, cambios en el color de la piel, aumento de la sudoración, daño muscular, dolor de espalda, dificultad para orinar, sangrado vaginal, impotencia sexual, períodos menstruales abundantes o ausentes, irritación vaginal, dolor de mamas, sofocos, escalofríos i, decoloración de la lengua, cambio en la percepción de olores, sed, trastornos dentales.
Temodal solo en el glioma que ha regresado o empeorado
Los siguientes efectos secundarios pueden ocurrir y pueden requerir atención médica.
Muy frecuentes (pueden afectar a más de 1 de cada 10 personas):
disminución del número de células sanguíneas (neutropenia o linfopenia, trombocitopenia), pérdida de apetito, dolor de cabeza, vómitos, náuseas (malestar estomacal), estreñimiento (dificultad para defecar), cansancio.
Frecuentes (pueden afectar hasta 1 de cada 10 personas):
pérdida de peso, somnolencia, mareos, sensación de hormigueo, dificultad para respirar, diarrea, dolor abdominal, malestar estomacal, erupción cutánea, picazón, caída del cabello, fiebre, debilidad, escalofríos, malestar, dolor, alteración del gusto.
Poco frecuentes (pueden afectar hasta 1 de cada 100 personas):
reducción del número de células sanguíneas (pancitopenia, anemia, leucopenia).
Raras (pueden afectar hasta 1 de cada 1.000 personas):
tos, infecciones que incluyen neumonía.
Muy raros (pueden afectar hasta 1 de cada 10.000 personas):
enrojecimiento de la piel, urticaria (ronchas), erupción cutánea, reacciones alérgicas.
Otros efectos secundarios:
Se han notificado con frecuencia casos de elevación de las enzimas hepáticas. Con poca frecuencia se han notificado casos de aumento de la bilirrubina, problemas con el flujo de bilis (colestasis), hepatitis y daño hepático, incluida insuficiencia hepática con resultado de muerte.
Se han observado casos muy raros de erupción grave con hinchazón de la piel, incluso en las palmas de las manos y plantas de los pies o enrojecimiento doloroso de la piel y / o ampollas en el cuerpo o en la boca. Informe a su médico de inmediato si ocurren estos casos.
Se han observado casos muy raros de efectos secundarios pulmonares con Temodal. Los pacientes suelen presentarse con dificultad para respirar y tos. Informe a su médico si nota alguno de estos síntomas.
En casos muy raros, los pacientes que toman Temodal y medicamentos similares pueden tener un pequeño riesgo de desarrollar cánceres secundarios, incluida leucemia.
Con poca frecuencia se han notificado infecciones por citomegalovirus nuevas o reactivadas (recurrentes) e infecciones por el virus de la hepatitis B reactivado.
Se han notificado con poca frecuencia casos de diabetes insípida. Los síntomas de la diabetes insípida incluyen la excreción de una gran cantidad de orina y sensación de sed.
Notificación de efectos secundarios
Si experimenta cualquier efecto adverso, consulte a su médico, farmacéutico o enfermero. Esto incluye cualquier posible efecto adverso que no se mencione en este prospecto. También puede notificar los efectos secundarios directamente a través del sistema nacional de notificación incluido en el Apéndice V.Efectos secundarios que puede ayudar proporcionar más información sobre la seguridad de este medicamento.
Caducidad y retención
Mantenga este medicamento fuera de la vista y del alcance de los niños, preferiblemente en un armario cerrado con llave. La ingestión accidental puede causar la muerte a los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta y la caja La fecha de caducidad se refiere al último día de ese mes.
Presentación botella
No conservar por encima de 30 ° C.
Guarde las cápsulas en el frasco original para protegerlas de la humedad.
Mantenga la botella bien cerrada.
Presentación en bolsita
No conservar por encima de 30 ° C.
Informe a su farmacéutico si nota algún cambio en el aspecto de las cápsulas.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que no necesita, ya que esto ayudará a proteger el medio ambiente.
Otra información
Qué contiene Temodal
- El principio activo es temozolomida.
- Temodal 5 mg cápsulas duras: cada cápsula contiene 5 mg de temozolomida.
- Temodal 20 mg cápsulas duras: cada cápsula contiene 20 mg de temozolomida.
- Temodal 100 mg cápsulas duras: cada cápsula contiene 100 mg de temozolomida.
- Temodal 140 mg cápsulas duras: cada cápsula contiene 140 mg de temozolomida.
- Temodal 180 mg cápsulas duras: cada cápsula contiene 180 mg de temozolomida.
- Temodal 250 mg cápsulas duras: cada cápsula contiene 250 mg de temozolomida.
Los demás componentes son:
- contenido de la cápsula: lactosa anhidra, sílice coloidal anhidra, almidón glicolato sódico tipo A, ácido tartárico, ácido esteárico (ver sección 2 "Temodal contiene lactosa").
- cáscara de la cápsula:
- Temodal 5 mg cápsulas duras: gelatina, dióxido de titanio (E 171), lauril sulfato de sodio, óxido de hierro amarillo (E 172), índigo carmín (E 132),
- Temodal 20 mg cápsulas duras: gelatina, dióxido de titanio (E 171), lauril sulfato de sodio, óxido de hierro amarillo (E 172),
- Temodal 100 mg cápsulas duras: gelatina, dióxido de titanio (E 171), lauril sulfato de sodio, óxido de hierro rojo (E 172),
- Temodal 140 mg cápsulas duras: gelatina, dióxido de titanio (E 171), lauril sulfato de sodio, índigo carmín (E 132),
- Temodal 180 mg cápsulas duras: gelatina, dióxido de titanio (E 171), lauril sulfato de sodio, óxido de hierro amarillo (E 172) y óxido de hierro rojo (E 172),
- Temodal 250 mg cápsulas duras: gelatina, dióxido de titanio (E 171), lauril sulfato de sodio. tinta de impresión: goma laca, propilenglicol, agua purificada, hidróxido de amonio, hidróxido de potasio y óxido de hierro negro (E 172).
Descripción del aspecto de Temodal y contenido del envase
- Las cápsulas duras de Temodal 5 mg tienen un cuerpo blanco opaco, una tapa verde opaca y están impresas con tinta negra.
- Las cápsulas duras de Temodal 20 mg tienen un cuerpo blanco opaco, una tapa amarilla opaca y están impresas con tinta negra.
- Las cápsulas duras de Temodal 100 mg tienen un cuerpo blanco opaco, una tapa rosa opaca y están impresas con tinta negra.
- Las cápsulas duras de Temodal 140 mg tienen un cuerpo blanco opaco, tapa azul y están impresas con tinta negra.
- Las cápsulas duras de Temodal 180 mg tienen un cuerpo blanco opaco, una tapa naranja opaca y están impresas con tinta negra.
- Las cápsulas duras de Temodal 250 mg tienen un cuerpo y una tapa de color blanco opaco y están impresas con tinta negra.
Presentación botella
Las cápsulas duras para uso oral están disponibles en frascos de vidrio ámbar que contienen 5 o 20 cápsulas.
La caja contiene una botella.
Presentación en bolsita
Las cápsulas duras para uso oral están disponibles en cajas que contienen 5 o 20 cápsulas duras, que se sellan individualmente en sobres.
Es posible que no se comercialicen todos los tamaños de envases.
Prospecto fuente: AIFA (Agencia Italiana de Medicamentos). Contenido publicado en enero de 2016. Es posible que la información presente no esté actualizada.
Para tener acceso a la versión más actualizada, es recomendable acceder al sitio web de la AIFA (Agencia Italiana de Medicamentos). Descargo de responsabilidad e información útil.
01.0 NOMBRE DEL MEDICAMENTO
CÁPSULAS DURAS TEMODAL 100 MG
02.0 COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada cápsula dura contiene 100 mg de temozolomida.
Excipiente con efectos conocidos:
Cada cápsula dura contiene 175,7 mg de lactosa anhidra.
Para consultar la lista completa de excipientes, ver sección 6.1.
03.0 FORMA FARMACÉUTICA
Cápsula dura (cápsula).
Las cápsulas duras tienen un cuerpo blanco opaco, una tapa rosa opaca y están impresas con tinta negra.
"Temodal" está impreso en la carcasa. "100 mg", el logotipo de Schering-Plough y dos rayas están impresas en el cuerpo.
04.0 INFORMACIÓN CLÍNICA
04.1 Indicaciones terapéuticas
Temodal está indicado en el tratamiento de:
• pacientes adultos con glioblastoma multiforme diagnosticado por primera vez en combinación con radioterapia (RT) y más tarde como monoterapia.
• pacientes pediátricos ≥ 3 años de edad, adolescentes y adultos con glioma maligno, como glioblastoma multiforme o astrocitoma anaplásico, que experimentan recaídas o progresión después del tratamiento estándar.
04.2 Posología y forma de administración
Temodal solo debe ser recetado por médicos con experiencia en el tratamiento oncológico de tumores cerebrales.
Se puede administrar terapia antiemética (ver sección 4.4).
Dosis
Pacientes adultos con glioblastoma multiforme diagnosticado por primera vez
Temodal se administra en combinación con radioterapia focal (fase concomitante) y posteriormente como monoterapia durante hasta 6 ciclos de temozolomida (TMZ) (fase de monoterapia).
Fase concurrente
TMZ se administra por vía oral a una dosis diaria de 75 mg / m2 durante 42 días en combinación con radioterapia focal (60 Gy administrados en 30 fracciones). No se recomiendan reducciones de dosis, pero en base a criterios de toxicidad hematológica y no hematológica, se tomarán decisiones semanales si retrasar o interrumpir la administración de TMZ. La administración de TMZ puede continuarse durante el período concomitante de 42 días (hasta un máximo de 49 días) si se cumplen todas las condiciones siguientes:
• recuento absoluto de neutrófilos (ANC) ≥ 1,5 x 109 / L
• recuento trombocítico ≥ 100 x 109 / l
• Criterios de toxicidad común (CTC) para toxicidad no hematológica ≤ Grado 1 (excepto para alopecia, náuseas y vómitos).
Se debe realizar un hemograma completo semanalmente durante el tratamiento. El tratamiento con TMZ debe suspenderse temporal o permanentemente durante la fase concomitante según los criterios de toxicidad hematológica y no hematológica indicados en la Tabla 1.
a: El tratamiento concomitante con TMZ puede continuarse cuando se cumplen todas las condiciones siguientes: recuento absoluto de neutrófilos ≥ 1,5 x 109 / L; recuento trombocítico ≥ 100 x 109 / L; Toxicidad CTC no hematológica ≤ Grado 1 (excepto para alopecia, náuseas, vómitos).
Fase de monoterapia
Cuatro semanas después del final de la fase concomitante de TMZ + RT, TMZ se administra hasta por 6 ciclos como monoterapia. La dosis del ciclo 1 (monoterapia) es de 150 mg / m2 una vez al día durante 5 días seguida de 23 días sin tratamiento. Al comienzo del ciclo 2, la dosis se aumenta a 200 mg / m2 si el CTC para la toxicidad no hematológica para el ciclo 1 es de grado ≤ 2 (excepto para alopecia, náuseas y vómitos), el recuento absoluto de neutrófilos (RAN) es ≥ 1,5 x 109 / ly el recuento de trombocitos es ≥ 100 x 109 / l. Si no se aumenta la dosis al ciclo 2, no se pueden realizar aumentos de dosis en los ciclos posteriores. Una vez aumentada, la dosis permanecerá en 200 mg / m2 por día durante los primeros 5 días de cada ciclo subsiguiente, a menos que se produzca toxicidad. Las reducciones de dosis y las interrupciones del tratamiento durante la fase de monoterapia deben realizarse de acuerdo con las Tablas 2 y 3.
Se debe realizar un hemograma completo el día 22 (21 días después de la primera dosis de TMZ) durante el tratamiento. Se debe reducir la dosis o interrumpir la administración de acuerdo con la Tabla 3.
Tabla 2. Niveles de dosis de TMZ en monoterapia
Tabla 3. Reducción o interrupción de la dosis de TMZ durante la monoterapia
a: Los niveles de dosis de TMZ se enumeran en la Tabla 2.
b: TMZ debe suspenderse si:
• el nivel de dosis -1 (100 mg / m2) todavía causa una toxicidad inaceptable
• La misma toxicidad no hematológica de Grado 3 sigue produciéndose después de la reducción de la dosis (excepto en el caso de alopecia, náuseas, vómitos).
Pacientes adultos y pediátricos de al menos 3 años de edad con glioma maligno recurrente o progresivo:
La terapia implica un curso de tratamiento de 28 días. En pacientes que no hayan recibido previamente quimioterapia, TMZ se administra por vía oral a una dosis de 200 mg / m2 una vez al día durante los primeros 5 días, seguida de una `` interrupción del tratamiento por un total de 28 días '' de 23 días. La dosis es de 150 mg / m2 una vez al día, que se aumentará en el segundo ciclo a 200 mg / m2 una vez al día durante 5 días en ausencia de toxicidad hematológica (ver sección 4.4).
Poblaciones especiales
Población pediátrica
En pacientes de 3 años de edad o mayores, TMZ solo debe usarse en glioma maligno recurrente o progresivo. La experiencia en estos niños es muy limitada (ver secciones 4.4 y 5.1) No se ha establecido la seguridad y eficacia de TMZ en niños menores de 3 años. No hay datos disponibles.
Pacientes con insuficiencia hepática o renal
La farmacocinética de TMZ en pacientes con función hepática normal es comparable a la de pacientes con insuficiencia hepática moderada o moderada. No se dispone de datos sobre la administración de TMZ en pacientes con insuficiencia hepática grave (clase C de Child) o insuficiencia renal. En base a las propiedades farmacocinéticas de TMZ, es poco probable que se requiera una reducción de la dosis en pacientes con insuficiencia hepática grave o cualquier grado de insuficiencia renal. Sin embargo, TMZ debe administrarse con precaución en estos pacientes.
Pacientes de edad avanzada
El análisis farmacocinético poblacional de pacientes de 19 a 78 años mostró que el aclaramiento de TMZ no se ve afectado por la edad. Sin embargo, en pacientes de edad avanzada (> 70 años) parece haber un mayor riesgo de neutropenia y trombocitopenia (ver sección 4.4).
Método de administración
Las cápsulas duras de Temodal deben tomarse con el estómago vacío.
Las cápsulas deben tragarse enteras con un vaso de agua y no deben abrirse ni masticarse.
Si se producen vómitos después de la administración de la dosis, no se puede administrar una segunda dosis el mismo día.
04.3 Contraindicaciones
Hipersensibilidad al principio activo oa alguno de los excipientes incluidos en la sección 6.1.
Hipersensibilidad a la dacarbazina (DTIC).
Mielosupresión grave (ver sección 4.4).
04.4 Advertencias especiales y precauciones de uso apropiadas
Infecciones oportunistas y reactivación de infecciones
Se han observado infecciones oportunistas (como neumonía por Pneumocystis jirovecii) y reactivación de infecciones (como VHB, CMV) durante el tratamiento con TMZ (ver sección 4.8).
Neumonía por Pneumocystis jirovecii
Se demostró que los pacientes que recibieron TMZ en combinación con RT en un estudio piloto después del programa de tratamiento extendido de 42 días tenían un riesgo particular de desarrollar neumonía. Pneumocystis jirovecii (PCP). En consecuencia, para todos los pacientes que reciben TMZ y RT de forma concomitante durante un régimen de 42 días (con un máximo de 49 días), independientemente del recuento de linfocitos, se requiere profilaxis de la PCP. Si se produce linfopenia, los pacientes deben continuar con la profilaxis hasta que la linfopenia haya regresado a Grado ≤ 1.
Se puede observar una recurrencia más amplia de la PCP cuando se administra TMZ en un régimen de dosis más prolongado. Sin embargo, todos los pacientes tratados con TMZ, especialmente aquellos que toman esteroides, deben ser monitoreados de cerca para el desarrollo de PCP independientemente del régimen de dosificación. Se han notificado casos de insuficiencia respiratoria mortal en pacientes que recibieron TMZ, especialmente en combinación con dexametasona u otros esteroides.
VHB
Se ha notificado hepatitis debida a la reactivación del virus de la hepatitis B (VHB), en algunos casos con desenlace fatal. Se debe consultar a expertos en enfermedades hepáticas antes de iniciar el tratamiento en pacientes con serología de hepatitis B positiva (incluidos aquellos con enfermedad activa). Los pacientes deben ser monitoreados y manejados adecuadamente durante el tratamiento.
Hepatotoxicidad
Se ha notificado lesión hepática, incluida insuficiencia hepática mortal, en pacientes tratados con TMZ (ver sección 4.8). Deben realizarse pruebas de función hepática basales antes de iniciar el tratamiento. Si los resultados son anormales, los médicos deben evaluar el beneficio / riesgo, incluida la posibilidad de insuficiencia hepática fatal, antes de comenzar con TMZ. Para los pacientes en un curso de tratamiento de 42 días, las pruebas de función hepática deben repetirse a la mitad del curso. Para todos los pacientes, se deben realizar pruebas de función hepática después de cada ciclo de tratamiento. Para los pacientes con insuficiencia hepática significativa, los médicos deben sopesar el beneficio / riesgo de continuar el tratamiento. La toxicidad hepática puede ocurrir varias semanas o más después del último tratamiento con temozolomida.
Neoplasias
También se han notificado muy raramente casos de síndrome mielodisplásico y neoplasias malignas secundarias, incluida la leucemia mieloide (ver sección 4.8).
Terapia antiemética
Las náuseas y los vómitos son muy comunes con TMZ.
Antes o después de la administración de TMZ, puede estar indicada la terapia antiemética.
Pacientes adultos con glioblastoma multiforme diagnosticado por primera vez
Se recomienda la profilaxis antiemética antes de la dosis inicial de la fase concomitante, mientras que se recomienda encarecidamente durante la fase de monoterapia.
Pacientes con glioma maligno recurrente o progresivo
En pacientes que hayan experimentado vómitos intensos (grado 3 o 4) en ciclos de tratamiento anteriores, puede ser necesaria una terapia antiemética.
Parámetros de laboratorio
La mielosupresión, incluida la pancitopenia prolongada, puede ocurrir en pacientes tratados con TMZ, lo que puede provocar anemia aplásica, lo que en algunos casos conduce a un desenlace fatal. En algunos casos, la exposición a medicamentos concomitantes asociados con anemia aplásica, incluyendo carbamazepina, fenitoína y sulfametoxazol / trimetoprima, complica la evaluación. Se deben evaluar los siguientes parámetros de laboratorio antes de la administración: RAN ≥ 1,5 x 109 / ly recuento de plaquetas ≥ 100 x 109 / L. Se debe realizar un hemograma completo y semanal el día 22 (21 días después de la primera administración) o dentro de las 48 horas hasta que el RAN sea> 1,5 x 109 / ly el recuento de plaquetas sea> 100 x 109 / l. Si el RAN se reduce a plaquetas es de 2, 150 mg / m2 y 200 mg / m2 La dosis mínima recomendada es de 100 mg / m2.
Población pediátrica
No existe experiencia clínica con el uso de TMZ en niños menores de 3 años. La experiencia clínica en niños mayores y adolescentes es muy limitada (ver secciones 4.2 y 5.1).
Pacientes de edad avanzada (> 70 años)
Los pacientes de edad avanzada parecen tener un mayor riesgo de neutropenia y trombocitopenia que los más jóvenes. Por tanto, TMZ debe administrarse con especial cuidado a los pacientes de edad avanzada.
Pacientes masculinos
Se debe advertir a los hombres en tratamiento con TMZ que no procreen hasta 6 meses después de la última dosis y que pregunten sobre la crioconservación de espermatozoides antes de iniciar el tratamiento (ver sección 4.6).
Lactosa
Este medicamento contiene lactosa. Los pacientes con intolerancia hereditaria a galactosa, insuficiencia de lactasa de Lapp o problemas de absorción de glucosa o galactosa no deben tomar este medicamento.
04.5 Interacciones con otros medicamentos y otras formas de interacción
En un estudio de fase I separado, la administración de TMZ con ranitidina no alteró la absorción de temozolomida ni la exposición a su metabolito activo monometil triazenoimidazol carboxamida (MTIC).
La administración de TMZ con alimentos da como resultado una disminución del 33% en la Cmáx y una disminución del 9% en el área bajo la curva (AUC).
Dado que no se puede excluir que el cambio en la Cmáx sea de importancia clínica, Temodal debe administrarse sin alimentos.
La evaluación farmacocinética poblacional de los estudios de fase II reveló que la coadministración de dexametasona, proclorperazina, fenitoína, carbamazepina, ondansetrón, antagonistas del receptor H2 o fenobarbital no alteró el aclaramiento de TMZ. La administración concomitante de ácido valproico se asocia con una disminución leve pero estadísticamente significativa del aclaramiento de TMZ.
No se han realizado estudios para determinar el efecto de TMZ sobre el metabolismo o la eliminación de otros medicamentos. Sin embargo, como TMZ no sufre metabolismo hepático y se caracteriza por una baja unión a proteínas, es poco probable que afecte a la farmacocinética de otros medicamentos ( ver sección 5.2).
El uso de TMZ en combinación con otros agentes mielosupresores puede aumentar la posibilidad de mielosupresión.
Población pediátrica
Los estudios de interacciones solo se han realizado en adultos.
04.6 Embarazo y lactancia
El embarazo
No hay datos sobre mujeres embarazadas. Se demostró toxicidad teratogénica y / o fetal en estudios preclínicos en ratas y conejos tratados con 150 mg / m2 de TMZ (ver sección 5.3). Temodal no debe administrarse a mujeres embarazadas. Si se considera el uso durante el embarazo, se debe informar a la paciente del riesgo potencial para el feto.
Hora de la comida
Se desconoce si TMZ se excreta en la leche materna; por lo tanto, se debe interrumpir la lactancia durante el tratamiento con TMZ.
Las mujeres en edad fértil
Se debe aconsejar a las mujeres en edad fértil que utilicen métodos anticonceptivos eficaces para evitar el embarazo mientras estén en tratamiento con TMZ.
Fertilidad masculina
TMZ puede tener efectos genotóxicos.Por lo tanto, se debe advertir a los hombres que reciben tratamiento con TMZ que no procreen hasta 6 meses después de la última dosis y que pregunten sobre la criopreservación de espermatozoides antes de iniciar el tratamiento debido a una posible infertilidad irreversible asociada con la terapia con TMZ.
04.7 Efectos sobre la capacidad para conducir y utilizar máquinas
TMZ tiene una influencia mínima sobre la capacidad para conducir y utilizar máquinas debido a la aparición de fatiga y somnolencia (ver sección 4.8).
04.8 Efectos indeseables
Experiencia de ensayos clínicos
En pacientes tratados con TMZ, ya sea en tratamiento concomitante con RT o como monoterapia después de RT para el glioblastoma multiforme recién diagnosticado, o como monoterapia en pacientes con glioma recidivante o progresivo, las reacciones adversas muy frecuentes notificadas son similares y son: náuseas, vómitos, estreñimiento. , anorexia, dolor de cabeza y fatiga. Las convulsiones se notificaron con mucha frecuencia en pacientes con glioblastoma multiforme diagnosticado por primera vez que recibieron monoterapia, y la erupción fue notificada con mucha frecuencia en pacientes con glioblastoma multiforme diagnosticado por primera vez que tomaron TMZ concomitantemente con RT y también como monoterapia y comúnmente en glioma recurrente. La mayoría de las reacciones adversas hematológicas se notifican como frecuentes o muy frecuentes en ambas indicaciones (Tablas 4 y 5); la frecuencia de los grados 3-4 de los valores de laboratorio se da después de cada tabla.
En las tablas, las reacciones adversas se clasifican según la clasificación de órganos del sistema y la frecuencia. Las clases de frecuencia se definen de acuerdo con las siguientes convenciones: Muy frecuentes (≥ 1/10); Frecuentes (≥ 1/100,
Diagnóstico precoz del glioblastoma multiforme
La Tabla 4 enumera las reacciones adversas que se produjeron durante el tratamiento en pacientes con glioblastoma multiforme recién diagnosticado durante las fases de tratamiento concomitante y en monoterapia.
* Un paciente que fue aleatorizado al grupo de solo RT recibió TMZ + RT.
Resultados de laboratorio
Se ha observado mielosupresión (neutropenia y trombocitopenia), que es una conocida toxicidad limitante de la dosis para la mayoría de los agentes citotóxicos, incluido TMZ. Cuando las anomalías de laboratorio se suman a las reacciones adversas durante la fase concomitante y la fase de monoterapia, en el 8% de los pacientes se observaron anomalías de neutrófilos de grado 3 o 4, incluidos acontecimientos neutropénicos. Se observaron cambios trombocíticos de grado 3 o 4, incluidos eventos trombocitopénicos, en el 14% de los pacientes que recibieron TMZ.
Glioma maligno recurrente o progresivo
En los estudios clínicos, las reacciones adversas relacionadas con el tratamiento más frecuentes fueron los trastornos gastrointestinales, a saber, náuseas (43%) y vómitos (36%). Estos eventos fueron generalmente de grado 1 o 2 (0 - 5 episodios de vómitos en 24 horas), autolimitados o controlados rápidamente por la terapia antiemética convencional. La incidencia de náuseas y vómitos intensos fue del 4%.
La Tabla 5 enumera las reacciones adversas encontradas durante los ensayos clínicos en el glioma maligno recidivante o progresivo y después de la comercialización de Temodal.
Resultados de laboratorio
Se produjo trombocitopenia y neutropenia de grado 3 o 4 en el 19% y el 17% de los pacientes tratados por glioma maligno, respectivamente. Esto dio lugar a la hospitalización y / o la interrupción del tratamiento con TMZ en el 8% y el 4% de los pacientes, respectivamente. La mielosupresión fue predecible (generalmente dentro de los primeros ciclos, con nadir entre el día 21 y el día 28), y la recuperación fue rápida generalmente dentro de 1 -2 semanas No se observó evidencia de mielosupresión acumulativa. La presencia de trombocitopenia puede aumentar el riesgo de hemorragia y la presencia de neutropenia o leucopenia que de infecciones.
Sexo
En un análisis farmacocinético poblacional de ensayos clínicos, hubo 101 mujeres y 169 hombres para los que se disponía de recuentos nadir de neutrófilos y 110 mujeres y 174 varones para los que se disponía de recuentos nadir de plaquetas. , y trombocitopenia (frente al 3%, en mujeres frente a hombres, en el primer ciclo de terapia). En un conjunto de datos de 400 sujetos con glioma recurrente, se produjo neutropenia de grado 4 en el 8% de las mujeres vs 4% de los hombres y trombocitopenia de grado 4 en el 8% de las mujeres vs 3% de los varones, en el primer ciclo de terapia. En un estudio de 288 sujetos con glioblastoma multiforme recién diagnosticado, se produjo neutropenia de grado 4 en el 3% de las mujeres. vs 0% de los hombres y trombocitopenia de grado 4 en el 1% de las mujeres vs 0% de varones, en el primer ciclo de terapia.
Población pediátrica
La TMZ oral se ha estudiado en pacientes pediátricos (de 3 a 18 años) con glioma de tronco encefálico recurrente o astrocitoma de alto grado recurrente, con un régimen de dosificación diario durante 5 días cada 28 días. Aunque los datos son limitados, se espera que la tolerancia en los niños sea similar a la de los adultos. No se ha establecido la seguridad de TMZ en niños menores de 3 años.
Experiencia poscomercialización
Se identificaron las siguientes reacciones adversas graves adicionales durante la exposición posterior a la comercialización:
† Incluidos los casos con desenlace fatal
* Frecuencias estimadas en base a estudios clínicos relevantes.
Notificación de sospechas de reacciones adversas
La notificación de sospechas de reacciones adversas que se produzcan tras la autorización del medicamento es importante, ya que permite un seguimiento continuo de la relación beneficio / riesgo del medicamento.Se invita a los profesionales sanitarios a notificar cualquier sospecha de reacciones adversas a través de la Agencia Italiana de Medicamentos. , sitio web: http://www.agenziafarmaco.gov.it/it/responsabili.
04.9 Sobredosis
Se han evaluado clínicamente en pacientes dosis de 500, 750, 1.000 y 1.250 mg / m2 (dosis total por ciclo durante 5 días). La toxicidad hematológica limitó la dosis y se notificó con cada dosis, pero se espera que sea más grave con dosis más altas. Un paciente recibió una sobredosis de 10.000 mg (dosis total de ciclo único, durante 5 días) y las reacciones adversas notificadas fueron pancitopenia, pirexia, insuficiencia multifuncional y muerte. Se han notificado casos de pacientes que tomaron la dosis recomendada durante más de 5 días (hasta 64 días) que notificaron efectos secundarios, incluida la ablación de la médula ósea, con o sin infección, en algunos casos graves y prolongados y que provocaron la muerte. En caso de sobredosis, se requiere una evaluación hematológica. Deben instituirse medidas de apoyo según sea necesario.
05.0 PROPIEDADES FARMACOLÓGICAS
05.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: Agentes antineoplásicos - Otros agentes alquilantes, código ATC: L01A X03.
Mecanismo de acción
La temozolomida es un triazeno que experimenta una rápida conversión química, a pH fisiológico, en el compuesto activo monometil triazenoimidazol carboxamida (MTIC). Se cree que la citotoxicidad de MTIC se debe principalmente a la alquilación en la posición O6 7 de la guanina con una alquilación adicional en la posición N. Se cree que las lesiones citotóxicas que se desarrollan posteriormente implican una reparación aberrante del aducto de metilo.
Eficacia clínica y seguridad
Diagnóstico precoz del glioblastoma multiforme
Un total de 573 pacientes fueron aleatorizados para recibir TMZ + RT (n = 287) o RT sola (n = 286). Los pacientes del grupo de TMZ + RT recibieron de forma concomitante TMZ (75 mg / m2) una vez al día, desde el primer día de RT hasta el último día de RT, durante 42 días (con un máximo de 49 días). La fase fue seguida de la administración. de TMZ en monoterapia (150-200 mg / m) en los días 1 a 5 de cada ciclo de 28 días, hasta un máximo de 6 ciclos, comenzando 4 semanas después del final de la RT. El brazo de control recibió solo RT. La profilaxis contra la neumonía fue necesario durante la RT y la terapia combinada con TMZ. Pneumocystis jirovecii (PCP).
TMZ se administró como terapia de rescate en la fase de seguimiento en 161 pacientes de los 282 (57%) en el brazo de RT sola y en 62 pacientes de los 277 (22%) en el brazo de TMZ + RT.
L"cociente de riesgo (HR) para la supervivencia general fue de 1,59 (IC del 95% para HR = 1,33 - 1,91) con un rango logarítmico p frente a 10%) es mayor en el brazo de RT + TMZ. La adición de TMZ concomitante a la RT, seguida de TMZ sola, en el tratamiento de pacientes con glioblastoma multiforme recién diagnosticado, demostró un aumento estadísticamente significativo en la supervivencia general (SG) en comparación con la RT sola.
Los resultados del estudio no fueron consistentes en el subgrupo de pacientes con un estado funcional bajo (OMS PS = 2, n = 70), en quienes la supervivencia general y el tiempo hasta la progresión fueron similares en ambos brazos. Sin embargo, no parece existir un nivel de riesgo inaceptable en este grupo de pacientes.
Glioma maligno recurrente o progresivo
Los datos de eficacia clínica en pacientes con glioblastoma multiforme progresivo o recidivante (estado funcional de Karnofsky [KPS] ≥ 70) después de la cirugía y la RT se obtuvieron en dos ensayos clínicos con TMZ oral. Uno realizado en 138 pacientes (el 29% de los cuales había recibido quimioterapia previamente) fue no comparativo y el otro, realizado con TMZ vs procarbazina en 225 pacientes (67% de los cuales habían recibido previamente quimioterapia basada en nitrosourea) fueron asignados al azar al control activo. En ambos estudios, el criterio principal de valoración fue la supervivencia libre de progresión (SLP) definida por resonancia magnética o el empeoramiento neurológico. En el estudio no comparativo, la SLP a los 6 meses fue del 19%, la mediana de la supervivencia libre de progresión fue de 2,1 meses y la mediana general la supervivencia fue de 5,4 meses La incidencia de respuesta objetiva (TRO) basada en la resonancia magnética fue del 8%.
En el estudio aleatorizado, controlado con activo, la SSP a los 6 meses fue significativamente mayor para TMZ que para la procarbazina (21% frente al 8%, respectivamente - ji cuadrado p = 0,008) con una mediana de SSP de 2,89 y 1,88, respectivamente. Meses ( prueba de rango logarítmico p = 0,0063). La mediana de supervivencia para TMZ y procarbazina fue de 7,34 y 5,66 meses, respectivamente (prueba de log rank p = 0,33). A los 6 meses, el porcentaje de pacientes que sobrevivieron fue significativamente mayor en el grupo de TMZ (60%) que en el grupo de procarbazina (44%) (ji cuadrado p = 0,019). Se observó un beneficio en pacientes previamente sometidos a quimioterapia con un KPS ≥ 80.
Los datos sobre el tiempo hasta el empeoramiento del estado neurológico fueron favorables para TMZ en comparación con la procarbazina, así como los datos sobre el tiempo hasta el empeoramiento del estado funcional (disminución de KPS en
Astrocitoma anaplásico recurrente
En un estudio prospectivo multicéntrico de fase II que evaluó la seguridad y eficacia de TMZ oral en el tratamiento de pacientes con astrocitoma anaplásico en la primera recaída, la SSP a los 6 meses fue del 46%. La mediana de SSP fue de 5, 4 meses. Mediana de supervivencia general fue de 14,6 meses. La tasa de respuesta, basada en la evaluación del revisor central, fue del 35% (13 RC y 43 RP) para la población por intención de tratar (ITT) n = 162 Se notificó enfermedad estable en 43 pacientes. La supervivencia libre para la población ITT fue del 44% con una mediana de supervivencia libre de eventos de 4,6 meses; estos resultados son similares a los de la supervivencia libre de progresión. Para la población elegible para histología, los resultados de eficacia fueron similares. La respuesta o el mantenimiento de la progresión libre se asoció fuertemente con el mantenimiento o mejora de la calidad de vida.
Población pediátrica
La TMZ oral se ha estudiado en pacientes pediátricos (de 3 a 18 años) con glioma de tronco encefálico recurrente o astrocitoma de alto grado recurrente, con un régimen de dosificación diario durante 5 días cada 28 días. La tolerancia a TMZ fue similar en adultos.
05.2 Propiedades farmacocinéticas
TMZ se hidroliza espontáneamente a pH fisiológico principalmente en la forma activa, 3-metil- (triazen-1-il) imidazol-4-carboxamida (MTIC). MTIC se hidroliza espontáneamente a 5-amino-imidazol-4-carboxamida (AIC), un intermedio conocido en la biosíntesis de purinas y ácidos nucleicos, y a metilhidrazina, que se cree que es la forma alquilante activa. MTIC se debe principalmente a la alquilación de ADN principalmente en las posiciones O6 7 y N de la guanina. En cuanto al AUC de TMZ, la exposición a MTIC y AIC es de 2,4% y 23%, respectivamente. En vivo, t1 / 2 de MTIC fue similar a la de TMZ, e igual a 1.8 h.
Absorción
Después de la administración oral en pacientes adultos, TMZ se absorbe rápidamente, alcanzando concentraciones máximas tan pronto como 20 minutos después de la dosis (tiempos medios entre 0,5 y 1,5 horas). Después de la administración oral de TMZ marcado con 1414C, la excreción fecal media de C durante los 7 días posteriores a la dosis fue del 0,8%, lo que demuestra una absorción completa.
Distribución
TMZ se caracteriza por una baja tendencia a unirse a proteínas (10% a 20%) y, por lo tanto, no se espera que interactúe con agentes que se unen fuertemente a proteínas.
Los estudios de PET en humanos y los datos preclínicos sugieren que TMZ atraviesa rápidamente la barrera hematoencefálica y está presente en el LCR. Se confirmó la penetración del LCR en un paciente; la exposición al LCR calculada en base al AUC. De TMZ, fue aproximadamente el 30% de la del plasma. coherente con los datos sobre animales.
Eliminación
La vida media (t1 / 2) en plasma es de aproximadamente 1.8 horas. La principal vía de eliminación del 14C es a través del riñón. Después de la administración oral aproximadamente 5% - 10% de la dosis se recupera sin cambios en la orina en 24 horas. horas y el resto se excreta como ácido temozolomida, 5-aminoimidazol-4-carboxamida (AIC) o como metabolitos polares no identificados.
Las concentraciones plasmáticas aumentan en función de la dosis. El aclaramiento plasmático, el volumen de distribución y la semivida son independientes de la dosis.
Poblaciones especiales
El análisis farmacocinético poblacional mostró que el aclaramiento plasmático de TMZ era independiente de la edad, la función renal y el consumo de tabaco.En un estudio farmacocinético separado, los perfiles farmacocinéticos plasmáticos en pacientes con insuficiencia hepática leve a moderada fueron similares a los observados en pacientes con función hepática normal.
Los pacientes pediátricos tenían un AUC más alto que los pacientes adultos; sin embargo, la dosis máxima tolerada (MDT) fue de 1.000 mg / m2 por ciclo tanto en niños como en adultos.
05.3 Datos preclínicos sobre seguridad
Se realizaron estudios de toxicidad de ciclo único (5 días de tratamiento y 23 días sin tratamiento), 3 y 6 ciclos en ratas y perros. Los objetivos de toxicidad primarios incluyeron la médula ósea, el sistema linforreticular, los testículos y el tracto gastrointestinal, y en dosis más altas, que fueron fatales en el 60% -100% de las ratas y perros analizados, se produjo degeneración de la retina. La mayoría de los efectos tóxicos fueron reversibles, excepto los eventos adversos que afectaron al sistema reproductor masculino y la degeneración de la retina. Sin embargo, dado que las dosis que conducen a la degeneración de la retina se encuentran en el rango de dosis letales y no se han observado efectos comparables en los ensayos clínicos, no se ha atribuido relevancia clínica a este hallazgo.
TMZ es un agente alquilante embriotóxico, teratogénico y genotóxico. TMZ es más tóxico en ratas y perros que en humanos, y la dosis clínica se acerca a la dosis letal más baja para ratas y perros. La reducción relacionada con la dosis de leucocitos y plaquetas parece ser un indicador significativo de toxicidad. En el estudio de 6 estudios En ciclos de ratas, se observaron varias neoplasias incluyendo carcinoma de mama, queratoacantoma de la piel, adenoma de células basales, mientras que en los estudios con perros no se observaron tumores ni cambios preneoplásicos. Las ratas parecen ser particularmente sensibles a los efectos oncogénicos de TMZ. Los primeros tumores aparecen dentro de los 3 meses desde el inicio de la administración. Este período de latencia también es muy corto para un agente alquilante.
Los resultados de la prueba de Ames / Salmonella y la prueba de aberración cromosómica de linfocitos de sangre periférica humana (HPBL) mostraron una respuesta de mutagenicidad positiva.
06.0 INFORMACIÓN FARMACÉUTICA
06.1 Excipientes
Contenido de la cápsula:
lactosa anhidra,
sílice coloidal anhidra,
glicolato sódico de almidón tipo A,
ácido tartárico,
ácido esteárico.
Carcasa de la cápsula:
gelatina,
dióxido de titanio (E 171),
Lauril Sulfato de Sodio,
óxido de hierro rojo (E 172)
Tinta de impresión:
goma laca,
propilenglicol,
agua purificada,
hidróxido de amonio,
hidróxido de potasio,
óxido de hierro negro (E 172).
06.2 Incompatibilidad
Irrelevante.
06.3 Período de validez
3 años
06.4 Precauciones especiales de conservación
Presentación botella
No conservar por encima de 30 ° C.
Guarde las cápsulas en el frasco original para protegerlas de la humedad.
Mantenga la botella bien cerrada.
Presentación en bolsita
No conservar por encima de 30 ° C.
06.5 Naturaleza del envase primario y contenido del envase.
Presentación botella
Frascos de vidrio ámbar tipo I con cierre de polipropileno a prueba de niños que contienen 5 o 20 cápsulas duras.
La caja contiene una botella.
Presentación en bolsita
Los sobres están compuestos de polietileno lineal de baja densidad (capa más interna), aluminio o tereftalato de polietileno.
Cada sobre contiene 1 cápsula dura y se suministra en una caja de cartón.
La caja contiene 5 o 20 cápsulas duras, en sobres sellados individualmente.
Es posible que no se comercialicen todos los tamaños de envases.
06.6 Instrucciones de uso y manipulación
No abra las cápsulas. Si una cápsula está dañada, evite el contacto del polvo que contiene con la piel o las membranas mucosas. Si hay contacto con la piel o las membranas mucosas, lave inmediata y minuciosamente el área afectada con agua y jabón.
Se debe advertir a los pacientes que mantengan las cápsulas fuera de la vista y del alcance de los niños, preferiblemente en un armario cerrado con llave. La ingestión accidental puede ser fatal para los niños.
Los medicamentos no utilizados y los desechos derivados de este medicamento deben eliminarse de acuerdo con las regulaciones locales.
07.0 TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Merck Sharp & Dohme Limited
Hertford Road, Hoddesdon
Hertfordshire EN11 9BU
Reino Unido
08.0 NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU / 1/98/096/005
EU / 1/98/096/006
EU / 1/98/096/015
EU / 1/98/096/016
034527059
034527061
034527150
034527162
09.0 FECHA DE LA PRIMERA AUTORIZACIÓN O RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 26 de enero de 1999
Fecha de la renovación más reciente: 26 de enero de 2009
10.0 FECHA DE REVISIÓN DEL TEXTO
27 de mayo de 2015