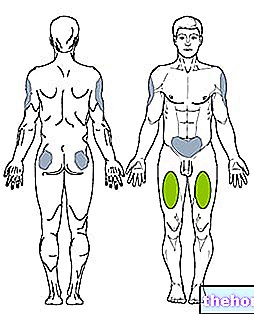
La lipoproteína lipasa (LPL) es una enzima que se encuentra en las células endoteliales que recubren la superficie interna de los capilares sanguíneos. Se concentra particularmente a nivel del endotelio capilar del tejido muscular esquelético, tejido cardíaco y tejido adiposo. No es de extrañar que la función de la lipoproteína lipasa sea hidrolizar los triglicéridos contenidos en las lipoproteínas (quilomicrones y VLDL), liberando dos ácidos grasos libre y un monoacilglicerol. Los productos derivados de esta hidrólisis de triglicéridos se difunden en las células, donde pueden cumplir esencialmente dos destinos: el primero es metabolizarse en el músculo esquelético y en el corazón, el segundo, propio de los períodos de sobrealimentación (excedente de energía), se utilizará como sustrato para la resíntesis de triglicéridos y luego se acumulará como reserva de energía.
La insulina aumenta la expresión de la lipoproteína lipasa a nivel del tejido adiposo blanco, favoreciendo la hidrólisis de los triglicéridos sanguíneos en glicerol y ácidos grasos, por lo que estos últimos pueden ingresar a los adipocitos y luego reesterificarse con glicerol, formando reserva de triglicéridos.
Deficiencia familiar de lipoproteína lipasa (enfermedad de Burger-Grutz o hiperlipoproteinemia familiar tipo I)
Enfermedad autosómica recesiva, con una incidencia igual a un caso por cada 100.000 personas. Ocurre en sujetos homocigotos para una mutación en el gen que codifica la lipoproteína lipasa. La consecuente deficiencia de esta enzima hace que los afectados por esta enfermedad presenten niveles particularmente elevados de triglicéridos (generalmente superiores a 800-1000 mg / dL), debido al bloqueo del metabolismo de los quilomicrones. La hipertrigliceridemia severa se acompaña, desde la infancia, de una mayor incidencia de pancreatitis, dolor abdominal, xantomas eruptivos (pápulas amarillentas con contornos enrojecidos distribuidos en las regiones corporales sometidas a presión) y hepatoesplenomegalia (agrandamiento anormal del hígado y bazo). riesgo cardiovascular, mientras que a veces se presenta retinopatía.
Deficiencia familiar de APO-C2
Una proteína muy importante, porque es capaz de activar la lipoproteína lipasa, es la Apo-lipo-proteína-C2 o APO-C2. Una deficiencia de esta proteína, expresada en la superficie de VLDL y quilomicrones, puede causar hiperlipoproteinemia caracterizada por hipertrigliceridemia (triglicéridos altos en sangre). En consecuencia, la deficiencia de APO-C2 se asocia con un mayor riesgo de aterosclerosis temprana y de pancreatitis, más común en ancianos. la edad. También en este caso la enfermedad está ligada a una mutación autosómica recesiva, específicamente en el gen que codifica APO-C2.
Lipoproteinlipasas, dieta, medicamentos y suplementos.
La deficiencia de lipoproteína lipasa o APO-C2 se puede tratar con una dieta baja en grasas, consumida en cantidades que no excedan los 10-20 gramos por día. Las grasas de cadena intermedia, que se unen directamente a la albúmina y no aprovechan los quilomicrones para ser transportadas en el torrente sanguíneo, deben ser claramente preferidas. Al mismo tiempo, es necesario eliminar el alcohol y asegurar un suministro adecuado de vitaminas y ácidos grasos liposolubles. esencial. Los omega-3 de origen pescado (EPA y DHA), en particular, han mostrado notables propiedades reductoras de hipotiglicéridos y, como tales, se utilizan en dosis altas para reducir los niveles de triglicéridos. Otros fármacos con actividad similar son los fibratos y el ácido nicotínico, que ejercen su acción reductora de hipotriglicéridos también aumentando la expresión de la enzima lipoproteína lipasa.