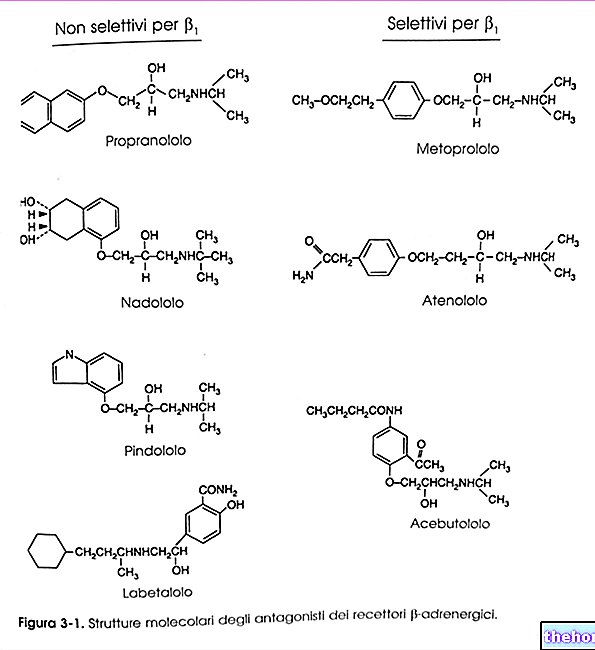
Esta enfermedad debe su nombre al endocrinólogo estadounidense que la descubrió: Frederic Crosby Bartter, cuya incidencia anual se ha estimado en 1 / 830.000.
Existen varias variantes del síndrome de Bartter cuya transmisión, aunque todavía autosómica, puede variar de recesiva a dominante según el caso.
Si no se diagnostica y trata con prontitud, el síndrome de Bartter puede comprometer gravemente el desarrollo, el crecimiento y la calidad de vida del paciente. Además, en casos particularmente graves, la esperanza de vida se reduce considerablemente.
Tenga en cuenta
El síndrome de Bartter NO debe confundirse con el síndrome de Schwartz-Bartter, una enfermedad caracterizada por "secreción alterada de hormona antiduiretica (ADH), también conocida como síndrome de secreción inapropiada de ADH (SIADH).
.que ocurre a nivel del asa de Henle es atribuible a una "alteración de la síntesis de algunos canales / receptores transportadores (proteínas particulares que transportan iones de diferente naturaleza) ubicados en esta" zona del riñón. Este fenómeno se produce por una serie de mutaciones genéticas que afectan a los genes que codifican las proteínas particulares antes mencionadas.
Las diferentes variantes del síndrome de Bartter se distinguen según el gen afectado. Puede encontrar información más detallada sobre esto en el siguiente capítulo.
.
Por tanto, la siguiente tabla mostrará las diferentes variantes del síndrome, los genes mutados implicados, las proteínas (canales receptores / transportadores) que codifican y la presentación clínica de la variante en cuestión.
Variante
Gen mutado
Canal / transportador involucrado
Presentación clínica
Síndrome de Bartter tipo I
Gen SLC12A1
NKCC2 (cotransportador de sodio-potasio-cloro o Na + / K + / 2Cl-)
Síndrome de Bartter prenatal (o infantil)
Síndrome de Bartter tipo II
Gene KCNJ1
ROMK (canal de potasio de la médula externa del riñón)
Síndrome de Bartter prenatal (o infantil)
Síndrome de Bartter tipo III
Gene CLNKb
CLCNKb (canal de cloro tipo Kb)
Síndrome de Bartter clásico
Síndrome de Bartter tipo IV o IV A
Gen BSND
Barttina (subunidad beta de canales de cloro tipo Ka y Kb)
Síndrome de Bartter prenatal (o infantil) e hipoacusia neurosensorial
Síndrome de Bartter tipo IV B
Genes CLCNKa y CLCNKb
CLCNKa (canal de cloro tipo Ka) y CLCNKb
Síndrome de Bartter prenatal (o infantil) y sordera neurosensorial
Síndrome de Bartter tipo V
Gen CASR
CaSR (receptor sensible al calcio)
Síndrome de Bartter con hipocalcemia
Como puede verse en la tabla, a pesar de la presencia de cinco variantes genéticas, no es posible distinguir tantas formas clínicas; de hecho, solo se distinguen cuatro: síndrome de Bartter prenatal o infantil (tipo I y II), síndrome de Bartter clásico (tipo III), síndrome de Bartter prenatal o infantil asociado a sordera neurosensorial (tipo IV A y IV B; algunas fuentes, sin embargo, agrupan estas variantes junto con el tipo I y II) y, finalmente, el síndrome de Bartter con hipocalcemia (tipo V).
Sabía usted que ...
Dada la existencia de una variante IV (o IV A) y una variante IV B del síndrome de Bartter, algunas fuentes consideran, en general, seis variantes del síndrome de Bartter. Sin embargo, otras fuentes consideran la variante IV B como un subtipo de la variante IV y , por ello, contemplan la existencia de solo cinco variantes genéticas del síndrome de Bartter.
Las variantes de los tipos I, II, III, IV y IV B son enfermedades de transmisión autosómica recesiva, lo que significa que para manifestar el síndrome el individuo debe poseer ambos alelos mutados heredando de los padres que, por tanto, serán portadores sanos. Variante V del síndrome, por otro lado, es una enfermedad de transmisión autosómica dominante, esto significa que para manifestar los síntomas, es suficiente que el paciente posea un solo alelo mutado que, por lo tanto, puede ser heredado incluso por uno solo (también enfermo ) de los dos padres.
Pseudo-síndrome de Bartter
El pseudo-síndrome de Bartter es una condición caracterizada por síntomas similares a los inducidos por el síndrome de Bartter pero cuya causa se encuentra en el abuso de fármacos diuréticos como la furosemida.
Síndrome de Gitelman
Este síndrome es causado por una mutación localizada en el gen SLC12A3 que codifica el cotransportador de sodio-cloro (NCC). Debido a esta mutación - transmitida de manera autosómica recesiva - el paciente sufre un deterioro de la reabsorción de sodio, cloro y potasio a nivel del túbulo contorneado distal, a diferencia del síndrome de Bartter en el que el deterioro de la reabsorción se localiza en el "Sin embargo , El síndrome de Gitelman puede dar lugar a síntomas similares a los del síndrome de Bartter, por lo que, en la práctica clínica, a veces puede resultar difícil distinguir las dos enfermedades.
, hipocloremia y alcalosis metabólica que pueden estar asociadas con hiperreninemia (aumento de renina en sangre) e hiperaldosteronismo. Claramente, todas estas condiciones pueden, a su vez, dar lugar a una serie de síntomas capaces de comprometer la calidad de vida del paciente (por ejemplo, náuseas, vómitos, mareos, debilidad, dolor de cabeza, hipotensión, etc.).
Además de lo dicho hasta ahora, cada variante puede dar lugar a manifestaciones y síntomas específicos estrechamente relacionados con el gen mutado y a la consiguiente implicación del canal o cotransportador que codifica este gen. Por lo tanto, los síntomas y manifestaciones típicos asociados con cada una de las cinco formas diferentes del síndrome de Bartter se describirán brevemente a continuación.
Síndrome de Bartter tipo I
En el síndrome de Bartter tipo I las mutaciones afectan el gen que codifica el cotransportador sodio-potasio-cloro presente en el asa de Henle, debido a la reabsorción comprometida se produce hipovolemia por pérdida de sales. Al mismo tiempo, dado que la reabsorción de calcio también está ligada a la actividad del cotransportador antes mencionado, estamos asistiendo al inicio de la hipercalciuria, todo lo cual puede conducir al inicio de la nefrocalcinosis. También es posible experimentar hipermagnesuria. Puede desarrollarse polihidramnios secundario a poliuria fetal en el período prenatal.
Síndrome de Bartter tipo II
El síndrome de Bartter tipo II es causado por una mutación en el gen que codifica el canal de potasio de la médula suprarrenal. Las manifestaciones y síntomas son similares a los de la variante I y también en este caso se puede encontrar polihidramnios secundario a poliuria fetal. Sin embargo, en una etapa temprana, el recién nacido puede experimentar acidosis metabólica hiperpotasémica transitoria. Esta condición luego evoluciona hacia el cuadro clínico característico del síndrome de Bartter.
Síndrome de Bartter tipo III
También conocido como síndrome de Bartter clásico, la variante III de la enfermedad es causada por mutaciones en el gen que codifica el canal de cloro de tipo Kb. Dado que los canales de cloro de tipo Ka se conservan en esta forma, los síntomas tienden a ser más leves, aunque todavía presentes. Generalmente no hay nefrocalcinosis.
Síndrome de Bartter tipo IV y IV B
En ambos tipos de variante IV existe la implicación de genes implicados en la correcta síntesis de los canales de cloro Ka y Kb. Dado que ambos canales están comprometidos, los síntomas tienden a ser más graves que en la variante III del síndrome. Los lactantes pueden presentar inicialmente un cuadro clínico que imita el hipoaldosteronismo pero que luego evoluciona hacia alcalosis metabólica hipopotasémica cuando el organismo intenta compensar la falta de actividad de los canales de calcio antes mencionados. La característica de las variantes IV y IV B del síndrome de Bartter es la aparición de sordera.
Síndrome de Bartter tipo V
La variante V del síndrome de Bartter está provocada por una mutación que afecta al gen que codifica el receptor sensible al calcio, involucrado en la inhibición de la reabsorción de agua y de varios iones, como calcio, potasio y sodio. El receptor da lugar a la aparición de hipocalcemia y hipercalciuria consecuente asociada con los síntomas característicos del síndrome de Bartter.
Sabía usted que ...
Las variantes I, II, IV y IV B del síndrome de Bartter, así como con el nombre de síndrome de Bartter prenatal, a veces también se denominan síndrome de hipeprostaglandina E2, ya que se caracterizan por un aumento de los niveles plasmáticos de esta prostaglandina.
- destinado a identificar la presencia y concentración de electrolitos (sodio, potasio, cloruro, magnesio, bicarbonato, calcio) y sustancias específicas (renina y aldosterona) en plasma y / o orina.Sin embargo, el diagnóstico definitivo solo es posible con la ejecución de pruebas genéticas específicas.
El diagnóstico diferencial, en cambio, debe situarse frente al pseudo-síndrome de Bartter, el síndrome de Gitelman, la fibrosis quística y la enfermedad celíaca.
En los casos en los que existe cierto riesgo (por ejemplo, padres con portadores sanos y / o enfermos) de que el recién nacido pueda manifestar la enfermedad, también es posible el diagnóstico prenatal.
de:
- Suplementos de sales minerales (en particular, pero no exclusivamente, potasio) para compensar la falta de reabsorción;
- Fármacos antiinflamatorios no esteroideos (AINE) como, por ejemplo, indometacina, que se administran con el objetivo de disminuir niveles excesivamente elevados de prostaglandina E2;
- Diuréticos ahorradores de potasio (administrados para reducir la excreción de potasio en la orina).
En los casos más graves y / o en condiciones estresantes (aparición de otras enfermedades, intervenciones quirúrgicas, etc.), la reposición de potasio y otras sales minerales se puede realizar por vía intravenosa, por supuesto, una operación similar debe ser realizada por sanitarios. personal especializado.