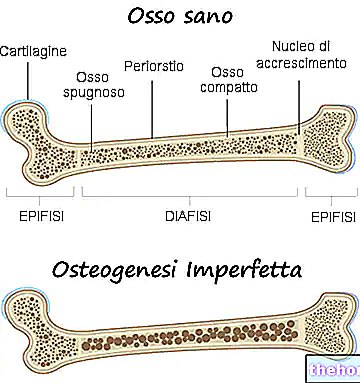
Generalidad
La osteogénesis imperfecta es una enfermedad genética congénita, no ligada al sexo, responsable de una cierta fragilidad ósea y una marcada tendencia a las fracturas.
Los síntomas de la osteogénesis imperfecta son numerosos; generalmente consisten en: debilitamiento óseo, alta tendencia a fracturas óseas, presencia de escleróticas oculares azules, grises o violáceas, presencia de deformidades óseas u otras alteraciones esqueléticas, cara triangular, fragilidad dentaria, etc. .
En general, los siguientes son esenciales para un correcto diagnóstico de osteogénesis imperfecta: examen físico, historial médico, pruebas de imagen médica, una prueba de evaluación de colágeno tipo I y una prueba genética.
Desafortunadamente, en la actualidad, los únicos tratamientos disponibles para los pacientes con osteogénesis imperfecta son sintomáticos. La enfermedad en cuestión, de hecho, es incurable.
¿Qué es la osteogénesis imperfecta?
La osteogénesis imperfecta es una enfermedad genética que debilita los huesos de la persona afectada y los hace más propensos a sufrir fracturas.
En realidad, con el término osteogénesis imperfecta, los médicos se refieren a un grupo heterogéneo de enfermedades genéticas, caracterizadas por un cierto grado de fragilidad ósea. Por tanto, existen varias formas (o tipos) de osteogénesis imperfecta, algunas mucho más graves que otras.
ES UNA ENFERMEDAD CONGÉNITA
En las personas que la padecen, la osteogénesis imperfecta es una enfermedad presente desde el nacimiento, por lo que se puede definir, a todos los efectos, una enfermedad congénita.
¿ESTÁ RELACIONADO CON EL SEXO?
La osteogénesis imperfecta no es una enfermedad genética relacionada con el género, como la hemofilia o el síndrome de Klinefelter.
EPIDEMILOGIA
Según algunas investigaciones estadísticas, la incidencia de osteogénesis imperfecta sería igual a un caso cada 15.000-20.000 nacimientos. Esto significa que cada 15.000-20.000 recién nacidos tiene uno afectado por osteogénesis imperfecta.
Otros estudios estadísticos también han demostrado que la osteogénesis imperfecta afecta a hombres y mujeres por igual, y que no tiene preferencia por una población o grupo étnico en particular.
La esperanza de vida es un parámetro extremadamente variable, que depende de la forma de osteogénesis imperfecta.
Causas
La osteogénesis imperfecta casi siempre resulta de una alteración cualitativa y cuantitativa de la producción de colágeno tipo I.
El colágeno tipo I es fundamental para fortalecer los huesos y mantener sanos los tejidos conectivos que constituyen cartílagos, tendones, piel, esclerótica ocular, etc.
Por tanto, una alteración en la producción de colágeno tipo I afecta la fortaleza de los huesos y la buena salud de los tejidos conectivos presentes en el cuerpo humano.
¿QUÉ ALTERA LA PRODUCCIÓN DE COLÁGENO?
Una enfermedad genética es una condición que surge debido a una mutación de uno o más genes que componen el ADN celular.
En el caso de la osteogénesis imperfecta, las causas de esta última casi siempre se encuentran en la mutación de uno o ambos genes COL1A1 (ubicado en el cromosoma 17) y COL1A2 (ubicado en el cromosoma 7).
En condiciones normales, COL1A1 y COL1A2 regulan la producción normal de colágeno tipo I; en presencia de mutaciones a su cargo, fallan en su función reguladora.
Importante: ¿qué otros genes, si están mutados, causan la osteogénesis imperfecta?
Además de las mutaciones de COL1A1 y COL1A2, las mutaciones en los genes IFITM5, SERPINF1, CRTAP y LEPRE1 son causas potenciales de osteogénesis imperfecta.
Los genes antes mencionados cubren funciones diferentes de COL1A1 y COL1A2, por lo tanto, no controlan la producción de colágeno tipo I, pero aún tienen una "influencia en la fuerza y resistencia de los huesos del esqueleto humano".
¿QUÉ TIPO DE ENFERMEDAD GENÉTICA ES?
La osteogénesis imperfecta es una enfermedad genética autosómica.
El término autosómico, asociado con una enfermedad genética, indica que la condición en cuestión se debe a mutaciones genéticas basadas en cromosomas autosómicos y no sexuales.
Se recuerda a los lectores que el ser humano posee un conjunto cromosómico de 23 pares de cromosomas totales, de los cuales 22 pares son del tipo autosómico y solo un par es del tipo sexual. El par de cromosomas del tipo sexual afecta al sexo del individual.
La osteogénesis imperfecta tras mutaciones en COL1A1, COL1A2 e IFITM5 tiene todas las características de una enfermedad autosómica dominante, cuando se debe a mutaciones en los genes SERPINF1, CRTAP y LEPRE1, tiene las características de una enfermedad autosómica recesiva.
TIPOS
Actualmente, los médicos creen que hay 8 tipos (o formas) de osteogénesis imperfecta. Para distinguir los distintos tipos, decidieron utilizar la numeración romana, para ser precisos los primeros ocho números romanos.
La siguiente tabla muestra las 8 formas de osteogénesis imperfecta, las mutaciones que las causan y otras características.
Chico
Gen mutado
Tipo de enfermedad genética
LOS
COL1A1
Dominante autosómico
II
COL1A1 y COL1A2
Dominante autosómico
III
COL1A1 y COL1A2
Dominante autosómico
IV
COL1A1 y COL1A2
Dominante autosómico
V.
IFITM5
Dominante autosómico
USTED
SERPINF1
Autosómica recesiva
VII
CRTAP
Autosómica recesiva
VIII
LIEBRE 1
Autosómica recesiva
* N.B: obviamente, las mutaciones en COL1A1 y COL1A2, que provocan las cuatro primeras formas de osteogénesis imperfecta, son alteraciones genéticas con características ligeramente diferentes. De lo contrario, no tendría sentido distinguir uno del otro.
Síntomas, signos y complicaciones.
Todos los tipos de osteogénesis imperfecta son responsables de un debilitamiento de los huesos, por lo que la persona afectada por la enfermedad tiene una predisposición particular a las fracturas. El grado de debilitamiento de los huesos varía según la forma; para algunos de ellos, este debilitamiento es mayor que para otros.
Dicho esto, cabe señalar que cada forma de osteogénesis imperfecta tiene su propio cuadro sintomático, que para algunos puede recordar el cuadro sintomatológico de otras formas.
POSIBLES SÍNTOMAS Y SIGNOS
Los posibles síntomas y signos de la osteogénesis imperfecta incluyen:
- Presencia de malformaciones óseas;
- Presencia de un cuerpo corto y pequeño (destinado a ser un tronco);
- Problemas en las articulaciones (por ejemplo, articulaciones flojas);
- Debilidad muscular;
- Esclera ocular azul, violeta o gris;
- Cara triangular;
- Cofre de barril;
- Anomalías morfológicas de la columna vertebral;
- Fragilidad dental;
- Rechazo o pérdida total de la audición;
- Problemas respiratorios
- Problemas relacionados con la ausencia o falta de colágeno tipo 1.
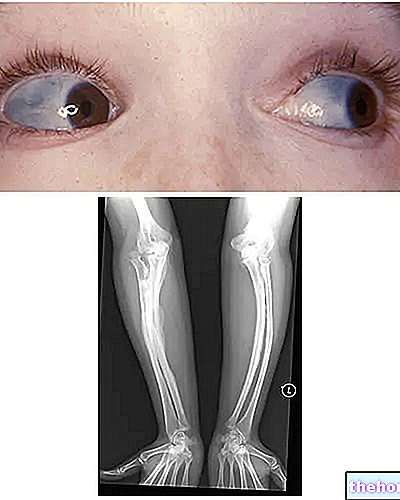
Osteogénesis imperfecta: nótese la coloración azul de las escleróticas y las deformaciones óseas que caracterizan la enfermedad. De wikipedia.org
¿CUÁLES SON LAS FORMAS MÁS GRAVES DE OSTEOGÉNESIS IMPERFECTA?
Los médicos clasifican la gravedad sintomatológica de los distintos tipos de osteogénesis imperfecta en una escala de 3 grados, que son: grado leve, grado moderado y grado severo.
Solo una forma pertenece a la categoría de "grado leve": "osteogénesis imperfecta tipo I"; 4 formas de osteogénesis imperfecta pertenecen a la categoría de "grado moderado": IV, V y VI; finalmente, a la categoría de "grado severo" pertenecen 3 formas: II, III, VII y VIII.
TIPO I: CARACTERÍSTICAS
La forma más común y menos grave de todas, la osteogénesis imperfecta tipo I tiene las siguientes características:
- Provoca fracturas especialmente antes de la pubertad;
- No tiene "casi ninguna influencia sobre la altura, por lo que los pacientes suelen tener una altura normal";
- Provoca problemas articulares y debilidad muscular.
- Es responsable de la esclerótica azul, violeta o gris;
- Es la causa de anomalías triangulares en la cara y la columna;
- Casi nunca causa deformidades óseas. Si los provoca, son mínimos;
- Puede provocar fragilidad dentaria y / o hipoacusia (esta última suele ocurrir en la edad adulta);
- Se asocia con la presencia de colágeno tipo I que es de calidad normal pero anormal en cantidad (es más pobre de lo normal).
TIPO II: CARACTERÍSTICAS
La osteogénesis imperfecta de tipo II se caracteriza por:
- Causa de muerte al nacer o poco tiempo después. Los problemas respiratorios casi siempre causan la muerte;
- Presencia de considerable fragilidad ósea y deformidades óseas graves;
- Estatura baja y pulmones subdesarrollados.
- Esclera de color azul, violeta o gris;
- Presencia de anomalías cuantitativas y cualitativas del colágeno tipo I.
TIPO III: CARACTERÍSTICAS
La osteogénesis imperfecta tipo III tiene las siguientes características:
- Aunque es muy grave, no suele causar la muerte en el período neonatal;
- Se asocia con "alta fragilidad ósea;
- Es responsable de baja estatura, problemas articulares, debilidad muscular (especialmente en piernas y brazos), pecho en forma de barril, cara triangular y curvatura anormal de la columna;
- Se debe a la esclerótica azul, violeta o gris;
- Puede causar problemas respiratorios, fragilidad dental y pérdida de audición;
- Con frecuencia es responsable de las deformidades óseas;
- Se asocia con anomalías cualitativas y cuantitativas del colágeno tipo I.
TIPO IV: CARACTERÍSTICAS
La osteogénesis tipo IV se caracteriza por:
- Un grado de fragilidad ósea entre las formas II y III y la forma I;
- Estatura más baja que la media;
- Esclera de color azul, violeta o gris;
- Deformidades óseas de entidad leve / moderada, anomalías leves de la columna y el tórax en forma de barril;
- Cara triangular;
- Posible presencia de fragilidad dental y pérdida auditiva;
- Presencia de anomalías del colágeno tipo I.
TIPO V: CARACTERÍSTICAS
La osteogénesis imperfecta de tipo V se parece a la osteogénesis imperfecta de tipo IV en algunos aspectos. Sin embargo, tiene algunas peculiaridades, que son:
- Esclera de color normal;
- Ausencia de fragilidad dental;
- Formación de callos óseos anormales, durante el proceso de curación de huesos fracturados;
- Calcificación de la membrana interósea que reside entre el radio y el cúbito. Esto perjudica la movilidad del antebrazo.
TIPO VI: CARACTERÍSTICAS
También la osteogénesis imperfecta tipo VI es similar a la forma IV, para distinguirla de esta última hay algunas peculiaridades, entre las que se encuentran los niveles elevados de fosfatasa alcalina en sangre y la presencia, en algunos huesos, de laminillas (óseas) similares a las espinas de los peces.
TIPO VII: CARACTERÍSTICAS
Sintomáticamente, la osteogénesis imperfecta de tipo VII puede parecerse al tipo IV en algunas circunstancias y al tipo II en otras.
Las peculiaridades de esta forma patológica grave incluyen:
- Baja estatura;
- La presencia de un húmero (hueso del brazo) y un fémur (hueso del muslo) extremadamente cortos;
- La presencia frecuente de una deformidad de la cadera, conocida como coxa vara.
TIPO VIII: CARACTERÍSTICAS
La osteogénesis imperfecta de tipo VIII recuerda mucho a las formas II y III.
Entre sus características peculiares destacan: el severo déficit de crecimiento, la severa hipomineralización esquelética y la ausencia (o escasa presencia) de la enzima prolil 3-hidroxilasa.
Diagnóstico
En general, el proceso de diagnóstico al que se someten los pacientes con sospecha de osteogénesis imperfecta comienza con un examen físico cuidadoso y una historia clínica cuidadosa; luego continúa, con un "análisis de los antecedentes familiares del paciente y con una serie de pruebas de diagnóstico por imagen (radiografías, tomografías computarizadas, etc.); finalmente, termina con una evaluación cuantitativa y cualitativa del colágeno tipo I y con una prueba genética.
Hoy en día, existe la posibilidad de diagnosticar osteogénesis imperfecta incluso en la fase prenatal, sometiendo a una mujer embarazada a una ecografía.
LA IMPORTANCIA DEL EXAMEN OBJETIVO Y LA HISTORIA
Un médico experto en osteogénesis imperfecta es capaz, muy a menudo, de diagnosticar la enfermedad antes mencionada incluso solo mediante un examen físico y una anamnesis. Esto significa que estas pruebas de diagnóstico no tienen una importancia despreciable.
EVALUACIÓN DE LA PRODUCCIÓN DE COLÁGENO TIPO I
Por regla general, la evaluación cualitativa y cuantitativa del colágeno tipo I es una prueba muy fiable, ya que, como se ha dicho, la mayoría de los casos de osteogénesis imperfecta se caracterizan por mutaciones en los genes que controlan la producción del colágeno tipo 1.
Para evaluar la cantidad y calidad del colágeno tipo I presente a nivel celular en un individuo, los médicos pueden confiar en una biopsia de piel o un análisis de sangre en particular.
Ambas pruebas de evaluación son bastante complejas y el paciente (o sus padres) pueden tener que esperar varias semanas para conocer los resultados.
PRUEBA GENÉTICA
Mediante una prueba genética que sondea todo el ADN del individuo examinado, los médicos pueden decretar definitivamente las características de la mutación genética presente.
Generalmente, se prevé la ejecución de un ensayo genético sobre todo el ADN celular cuando la evaluación de las características del colágeno tipo I no ha dado los resultados deseados, o cuando no es una mutación en COL1A1 o COL1A2 la que provoca la "osteogénesis imperfecta".
DIAGNÓSTICO PRENATAL
La ecografía prenatal es muy útil para identificar la osteogénesis imperfecta tipo II y tipo III.
Terapia
Actualmente no existe una cura específica para la osteogénesis imperfecta, es decir, las personas con osteogénesis imperfecta están destinadas a vivir con la afección antes mencionada hasta la muerte, que muchas veces se debe a las consecuencias de la propia enfermedad.
La falta de terapia específica no excluye la existencia de otras formas de tratamiento. De hecho, entre las posibilidades terapéuticas de un paciente con osteogénesis imperfecta se incluyen diversas terapias sintomáticas; Por terapias sintomáticas entendemos tratamientos capaces de aliviar los síntomas, ralentizar el curso de la enfermedad y prevenir (o al menos posponer) las consecuencias más graves.
POSIBLES TRATAMIENTOS SINTOMÁTICOS
En la lista de posibles tratamientos sintomáticos de la osteogénesis imperfecta destacan los siguientes:
- La inserción quirúrgica, en el interior de los huesos más largos (N.B: los más propensos a fracturas), de clavos que aporten mayor resistencia a las fracturas y deformidades. Esta operación se llama varillaje intramedular
- Tratamiento conservador o quirúrgico de fracturas y / o deformidades óseas;
- Cuidado dental, para salvaguardar la salud de los dientes;
- Terapias para aliviar el dolor, en caso de fracturas múltiples muy dolorosas;
- Fisioterapia, para el alargamiento y fortalecimiento muscular Un aparato muscular elástico y tónico permite prevenir caídas que pueden provocar diversas fracturas óseas;
- El uso de ayudas para la locomoción, incluidas sillas de ruedas, tirantes, muletas, etc.
BENEFICIOS DEL MOVIMIENTO
Para las personas con osteogénesis imperfecta, los médicos recomiendan la práctica constante de ejercicio físico y el movimiento en general, ya que ambas actividades contribuyen al fortalecimiento de los sistemas esquelético y muscular.
Entre los deportes recomendados se encuentran: la natación, ya que es una "actividad física de bajo impacto en el sistema esquelético", y la caminata.
BENEFICIOS DE UN ESTILO DE VIDA SALUDABLE
Llevar una vida sana, evitar fumar, beber alcohol en exceso, comer demasiado y mal, etc., tiene beneficios para la salud más que discretos para los pacientes con osteogénesis imperfecta, ya que ralentiza la progresión de la enfermedad y reduce la fragilidad ósea.
TRATAMIENTOS SINTOMÁTICOS EN LA FASE DE EXPERIMENTACIÓN
Actualmente, médicos e investigadores están evaluando la efectividad de algunos tratamientos sintomáticos, incluido el tratamiento con hormona del crecimiento y la terapia intravenosa y oral a base de bisfosfonatos.
Por el momento, los resultados proporcionados por los tratamientos de investigación antes mencionados son un buen augurio para toda la comunidad médica.
Pronóstico
La osteogénesis imperfecta es una enfermedad de pronóstico negativo, ya que es incurable, compromete drásticamente la calidad de vida y, en algunos casos, provoca la muerte prematura del sujeto afectado.
Sin embargo, cabe señalar que, también gracias a los tratamientos sintomáticos modernos, muchas personas con una forma leve de osteogénesis imperfecta pueden llevar una vida placentera y satisfactoria.
Prevención
Desafortunadamente, actualmente no existe una medida preventiva contra la osteogénesis imperfecta.





-cos-cause-e-terapia.jpg)