Ingredientes activos: Letrozol
Femara 2,5 mg comprimidos recubiertos con película.
¿Por qué se usa Femara? ¿Para qué sirve?
Qué es Femara y cómo funciona
Femara contiene un principio activo llamado letrozol. Pertenece a un grupo de medicamentos denominados inhibidores de la aromatasa y es un tratamiento hormonal (o "endocrino") para el cáncer de mama. El estrógeno, que es una hormona sexual femenina, estimula con frecuencia el crecimiento del cáncer de mama. Femara reduce la cantidad de estrógeno al bloquear una enzima ('aromatasa') que participa en la producción de estrógeno y, por lo tanto, puede bloquear el crecimiento de los tumores de mama que necesitan estrógeno para crecer. Como resultado, el crecimiento de las células cancerosas y / o su diseminación a otras partes del cuerpo se ralentiza o detiene.
Para qué se utiliza Femara
Femara se utiliza para tratar el cáncer de mama en mujeres posmenopáusicas que ya no tienen la menstruación.
Se utiliza para prevenir la reaparición del cáncer de mama. Se puede utilizar como primer tratamiento antes de la cirugía de mama si no es posible la cirugía inmediata o como primer tratamiento después de la cirugía de mama o después de cinco años de tratamiento con tamixofeno. Femara también se utiliza para prevenir la propagación del cáncer de mama a otras partes del cuerpo. en pacientes con cáncer de mama avanzado.
Si tiene alguna pregunta sobre cómo actúa Femara o por qué le han recetado este medicamento, consulte a su médico.
Contraindicaciones Cuándo no se debe usar Femara
Siga cuidadosamente las instrucciones de su médico. Pueden diferir de la información general proporcionada en este prospecto.
No tome Femara
- si es alérgico al letrozol oa cualquiera de los demás componentes de este medicamento
- si todavía está menstruando, es decir, si aún no está en la menopausia,
- sí estas embarazada,
- si está amamantando.
Si se encuentra en alguna de estas situaciones, no tome este medicamento e informe a su médico.
Precauciones de uso Lo que necesita saber antes de tomar Femara
Consulte a su médico o farmacéutico antes de tomar Femara.
- si tiene una enfermedad grave del riñón,
- si tiene una enfermedad grave del hígado,
- si tiene antecedentes de osteoporosis o fracturas óseas (ver también "Seguimiento del tratamiento con Femara" en la sección).
Si se encuentra en alguna de estas situaciones, informe a su médico. Su médico lo tendrá en cuenta durante el tratamiento con Femara.
Niños y adolescentes (menores de 18 años)
Los niños y adolescentes no deben usar este medicamento.
Ancianos (65 años o más)
Las mujeres de 65 años o más pueden usar este medicamento a la misma dosis que las mujeres adultas.
Interacciones ¿Qué medicamentos o alimentos pueden cambiar el efecto de Femara?
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tomar cualquier otro medicamento, incluso los adquiridos sin receta.
Advertencias Es importante saber que:
Embarazo, lactancia y fertilidad
- Solo debe tomar Femara cuando haya entrado en la menopausia.Sin embargo, su médico le explicará la necesidad de utilizar un sistema anticonceptivo eficaz, ya que podría quedar embarazada durante el tratamiento con Femara.
- No debe tomar Femara si está embarazada o amamantando, ya que puede dañar al bebé.
Conducción y uso de máquinas
Si se siente mareado, cansado, somnoliento o malestar general, no conduzca ni maneje maquinaria hasta que se sienta normal nuevamente.
Femara contiene lactosa
Femara contiene lactosa (azúcar de la leche). Si su médico le ha indicado que padece una intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento.
Para quienes realizan actividades deportivas: el uso de la droga sin necesidad terapéutica constituye dopaje y en cualquier caso puede determinar pruebas antidopaje positivas.
Dosis, método y momento de administración Cómo usar Femara: Posología
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico.
En caso de duda, consulte a su médico o farmacéutico. La dosis habitual es un comprimido de Femara que se debe tomar una vez al día. Tomar Femara a la misma hora cada día le ayudará a recordar cuándo tomar su comprimido.
La tableta debe tomarse con o sin comida y debe tragarse entera con un vaso de agua u otra bebida.
Cuánto tiempo tomar Femara
Continúe tomando Femara todos los días durante el tiempo que le haya indicado su médico. Es posible que deba tomarlo durante meses o incluso años. Si tiene alguna pregunta sobre cuánto tiempo debe tomar Femara, hable con su médico.
Seguimiento durante el tratamiento con Femara
Debe tomar este medicamento bajo la estricta supervisión de su médico. Su médico controlará su salud con regularidad para asegurarse de que el tratamiento tenga el efecto correcto.
Femara puede causar fragilidad o pérdida de masa ósea (osteoporosis) debido a una disminución de los estrógenos en el cuerpo. Su médico puede decidir que le midan la densidad ósea (una forma de comprobar la presencia de osteoporosis) antes, durante y después del tratamiento.
Sobredosis Qué hacer si ha tomado demasiado Femara
Si toma más Femara del que debiera
Si ha tomado demasiado Femara, o si otra persona accidentalmente toma sus comprimidos, póngase en contacto con su médico o con el hospital inmediatamente para que le aconsejen. Muéstreles el envase de los comprimidos. Puede que necesite tratamiento médico.
Si olvidó tomar Femara
- Si es casi la hora de su próxima dosis (por ejemplo, dentro de 2 o 3 horas), omita la dosis omitida y tome la siguiente dosis cuando se supone que debe hacerlo.
- De lo contrario, tome la dosis olvidada tan pronto como lo recuerde y luego tome la siguiente tableta como lo haría normalmente.
- No tome una dosis doble para compensar las dosis olvidadas.
Si deja de tomar Femara
No deje de tomar Femara a menos que su médico se lo indique Consulte también más arriba en "Cuánto tiempo debe tomar Femara".
Efectos secundarios ¿Cuáles son los efectos secundarios de Femara?
Como todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
La mayoría de los efectos secundarios son de leves a moderados y, por lo general, desaparecen después de un período de tratamiento que va desde unos pocos días hasta algunas semanas.
Algunos de estos efectos secundarios, como los sofocos, la caída del cabello o el sangrado vaginal, pueden deberse a la falta de estrógeno en el cuerpo.
No se preocupe por esta lista de posibles efectos secundarios. Puede que no esté sujeto a él.
Algunos efectos colaterales pueden ser serios:
Efectos raros o poco frecuentes (es decir, pueden afectar entre 1 y 100 pacientes de cada 10.000):
- Debilidad, parálisis o pérdida de sensibilidad en cualquier otra parte del cuerpo (especialmente brazo o pierna), pérdida de coordinación, náuseas o dificultad para hablar o respirar (síntoma de un trastorno cerebral como un accidente cerebrovascular).
- Dolor repentino y opresivo en el pecho (síntoma de enfermedad cardíaca).
- Dificultad para respirar, dolor en el pecho, desmayos, latidos cardíacos rápidos, coloración azul de la piel o dolor repentino en el brazo, la pierna o el pie (síntomas de una posible formación de coágulos de sangre).
- Hinchazón y enrojecimiento en correspondencia de una vena que es extremadamente sensible y también dolorosa al tacto.
- Fiebre alta, escalofríos o úlceras en la boca causadas por infecciones (falta de glóbulos blancos).
- Visión borrosa severa y persistente.
Si ocurre alguno de estos síntomas, informe a su médico de inmediato.
Debe informar a su médico de inmediato si experimenta alguno de los siguientes síntomas mientras está en tratamiento con Femara:
- Hinchazón principalmente de la cara y la garganta (signos de una reacción alérgica).
- Piel y ojos amarillentos, náuseas, pérdida del apetito, orina oscura (signos de hepatitis).
- Erupción, enrojecimiento de la piel, ampollas en los labios, ojos o labios, descamación de la piel, fiebre (signos de enfermedad de la piel).
Algunos efectos secundarios son muy frecuentes. Estos efectos secundarios pueden afectar a más de 10 de cada 100 pacientes.
- Sofocos
- Aumento de los niveles de colesterol (hipercolesterolemia).
- Fatiga
- Aumento de la sudoración
- Dolor en los huesos y las articulaciones (artralgia).
Si alguno de estos le afecta gravemente, informe a su médico.
Algunos efectos secundarios son frecuentes. Estos efectos secundarios pueden afectar entre 1 y 10 de cada 100 pacientes.
- Sarpullido
- Dolor de cabeza
- Mareo
- Malestar (generalmente malestar)
- Trastornos gastrointestinales como náuseas, vómitos, indigestión, estreñimiento, diarrea.
- Aumento o pérdida del apetito
- Dolor muscular
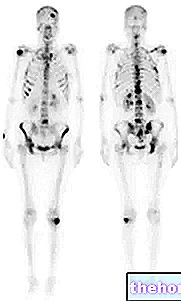
- Fragilidad o pérdida de masa ósea (osteoporosis), que en algunos casos conduce a fracturas óseas (ver también "Monitorización durante el tratamiento con Femara en la sección)
- Hinchazón de brazos, manos, pies, tobillos (edema).
- Depresión
- Aumento de peso
- Perdida de cabello
- Aumento de la presión arterial (hipertensión)
- Dolor abdominal
- Sequedad de la piel
- Sangrado vaginal
- Si alguno de estos le afecta gravemente, informe a su médico.
Otros efectos secundarios son poco frecuentes. Estos efectos secundarios pueden afectar entre 1 y 10 de cada 1000 pacientes.
- Trastornos del sistema nervioso como ansiedad, nerviosismo, irritabilidad, somnolencia, problemas de memoria, somnolencia, insomnio.
- Dolor o sensación de ardor en las manos o la muñeca (síndrome del túnel carpiano)
- Sensibilidad alterada, especialmente al tacto.
- Trastornos oculares como visión borrosa, irritación ocular.
- Palpitaciones, latidos cardíacos rápidos
- Trastornos de la piel como picazón (urticaria)
- Secreción o sequedad vaginal
- Rigidez articular (artritis)
- Dolor en los senos
- Fiebre
- Sed, alteraciones del gusto, sequedad de boca
- Sequedad de las membranas mucosas.
- Pérdida de peso
- Infecciones del tracto urinario, aumento de la frecuencia urinaria.
- Tos
- Aumento de los niveles de enzimas en el hígado.
Reacciones adversas con frecuencia no conocida.
Chasquear el dedo, una condición en la que uno de los dedos de la mano se atasca en una posición doblada.
Si alguno de estos le afecta gravemente, informe a su médico.
Si experimenta cualquier efecto adverso, consulte a su médico o farmacéutico, incluido cualquier posible efecto adverso no mencionado en este prospecto.
Notificación de efectos secundarios
Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluidos los posibles efectos adversos que no aparecen en este prospecto. También puede notificar efectos secundarios directamente a través del sistema nacional de notificación en https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse Al notificar efectos secundarios, puede ayudar a proporcionar más información sobre la seguridad de este medicamento.
Caducidad y retención
- Mantener fuera del alcance y de la vista de los niños.
- No utilice Femara después de la fecha de caducidad que aparece en la caja después de CAD. La fecha de caducidad se refiere al último día del mes.
- No conservar por encima de 30 ° C.
- Almacene en el paquete original para proteger el medicamento de la humedad.
- No utilice envases que estén dañados o muestren signos de alteración.
Qué contiene Femara
- El ingrediente activo es letrozol. Cada comprimido recubierto con película contiene 2,5 mg de letrozol.
- Los demás componentes son lactosa monohidrato, celulosa microcristalina, almidón de maíz, carboximetil almidón de sodio, estearato de magnesio y sílice coloidal anhidra. El recubrimiento se compone de hipromelosa, talco, macrogol 8000, dióxido de titanio (E 171) y óxido de hierro amarillo (E 172).
Aspecto de Femara y contenido del envase
- Femara se presenta en forma de comprimidos recubiertos con película. Los comprimidos recubiertos con película son de color amarillo oscuro y de forma redonda. Están marcados en un lado con "FV" y con "CG" en el otro lado.
- Cada blíster contiene 10, 14, 28, 30 o 100 comprimidos. Es posible que no todos los tamaños de envases estén disponibles en su país.
Prospecto fuente: AIFA (Agencia Italiana de Medicamentos). Contenido publicado en enero de 2016. Es posible que la información presente no esté actualizada.
Para tener acceso a la versión más actualizada, es recomendable acceder al sitio web de la AIFA (Agencia Italiana de Medicamentos). Descargo de responsabilidad e información útil.
01.0 NOMBRE DEL MEDICAMENTO
COMPRIMIDOS FEMARA 2.5 MG RECUBIERTOS CON PELÍCULA
02.0 COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
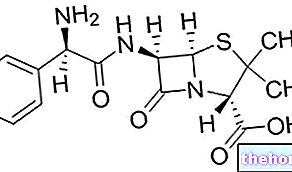
Principio activo: letrozol.
Cada comprimido recubierto con película contiene 2,5 mg de letrozol.
Cada comprimido contiene 61,5 mg de lactosa. Para consultar la lista completa de excipientes, ver sección 6.1.
03.0 FORMA FARMACÉUTICA
Comprimido recubierto con película.
Comprimido recubierto con película de color amarillo oscuro, redondo, ligeramente biconvexo con bordes biselados. Una parte lleva la inscripción "FV", la otra "CG".
04.0 INFORMACIÓN CLÍNICA
04.1 Indicaciones terapéuticas
Tratamiento adyuvante del cáncer de mama invasivo temprano en mujeres posmenopáusicas con estado positivo de receptores hormonales.
Tratamiento adyuvante del cáncer de mama invasivo hormonosensible en mujeres posmenopáusicas después del tratamiento adyuvante estándar con tamoxifeno durante 5 años.
Tratamiento de primera línea del cáncer de mama avanzado hormonosensible en mujeres posmenopáusicas.
Tratamiento del cáncer de mama avanzado en mujeres posmenopáusicas inducidas de forma natural o artificial después de la recurrencia o progresión de la enfermedad que han sido tratadas previamente con antiestrógenos.
Tratamiento neoadyuvante en mujeres posmenopáusicas con cáncer de mama HER-2 negativo con receptor hormonal positivo donde la quimioterapia no es posible y la cirugía inmediata no está indicada.
No se ha demostrado eficacia en pacientes con estado negativo de receptores hormonales.
04.2 Posología y forma de administración
Dosis
Pacientes adultos y ancianos
La dosis recomendada de Femara es de 2,5 mg una vez al día. No se requiere modificación de la dosis en pacientes de edad avanzada.
En pacientes con cáncer de mama avanzado o metastásico, se debe continuar el tratamiento con Femara hasta que sea evidente la progresión del tumor.
En el tratamiento adyuvante y el tratamiento adyuvante después de la terapia estándar con tamoxifeno, el tratamiento con Femara debe continuarse durante 5 años o hasta que ocurra la recurrencia del tumor, lo que ocurra primero.
También se puede considerar un régimen de tratamiento secuencial (letrozol durante 2 años seguido de tamoxifeno durante 3 años) como tratamiento adyuvante (ver secciones 4.4 y 5.1).
En el tratamiento neoadyuvante, el tratamiento con Femara debe continuarse durante 4 a 8 meses para establecer una reducción óptima del tumor. Si la respuesta es inadecuada, se debe interrumpir el tratamiento con Femara.
y se debe planificar la cirugía y / o se deben discutir con el paciente alternativas terapéuticas adicionales.
Población pediátrica
Femara no está recomendado para uso en niños y adolescentes No se ha establecido todavía la seguridad y eficacia de Femara en niños y adolescentes mayores de 17 años. Se dispone de datos limitados y no se puede hacer una recomendación posológica.
Insuficiencia renal
No se requiere modificación de la dosis de Femara en pacientes con insuficiencia renal con aclaramiento de creatinina ≥ 10 ml / min. Se dispone de datos insuficientes en casos de insuficiencia renal con aclaramiento de creatinina por debajo de 10 ml / min (ver secciones 4.4 y 5.2).
Deterioro hepático
No se requiere modificación de la dosis de Femara en pacientes con insuficiencia hepática leve a moderada (Child-Pugh A o B). Se dispone de datos insuficientes para pacientes con insuficiencia hepática grave. Los pacientes con insuficiencia hepática grave (Child-Pugh C) requieren una estrecha monitorización (ver secciones 4.4 y 5.2).
Método de administración
Femara debe tomarse por vía oral y puede tomarse con o sin alimentos.
La dosis olvidada debe tomarse tan pronto como el paciente se acuerde. Sin embargo, si es casi la hora de la siguiente dosis (dentro de 2 a 3 horas), no se debe tomar la dosis omitida y el paciente debe volver a su horario habitual de ingesta. Las dosis no deben duplicarse porque con dosis diarias superiores a la dosis recomendada de 2,5 mg, se observó una exposición sistémica sobreproporcionada (ver sección 5.2).
04.3 Contraindicaciones
Hipersensibilidad al principio activo oa alguno de los excipientes incluidos en la sección 6.1.
Estado hormonal premenopáusico
Embarazo (ver sección 4.6)
Lactancia materna (ver sección 4.6)
04.4 Advertencias especiales y precauciones de uso apropiadas
Estado menopáusico
En pacientes en las que el estado de la menopausia no está claro, se debe medir la hormona luteinizante (LH), la hormona estimulante del folículo (FSH) y / o el estradiol antes de iniciar el tratamiento con Femara. Solo las mujeres con el estado hormonal posmenopáusico pueden recibir Femara.
Insuficiencia renal
Femara no se ha estudiado en un número suficiente de pacientes con un aclaramiento de creatinina inferior a 10 ml / min. En estos pacientes, se debe considerar cuidadosamente la relación potencial beneficio / riesgo antes de la administración de Femara.
Deterioro hepático
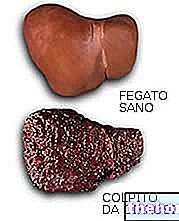
En pacientes con insuficiencia hepática grave (Child-Pugh C), la exposición sistémica y la semivida terminal son aproximadamente el doble que en voluntarios sanos. Por tanto, estos pacientes deben ser controlados de cerca (ver sección 5.2).
Efectos sobre el hueso
Femara es un poderoso agente reductor de estrógenos. Los pacientes con antecedentes de osteoporosis y / o fracturas, o con mayor riesgo de osteoporosis, deben someterse a una evaluación de la densidad mineral ósea antes de iniciar el tratamiento adyuvante y adyuvante después del tratamiento estándar con tamoxifeno y deben ser controlados durante y después del tratamiento con letrozol. Tratamiento o profilaxis.
La osteoporosis debe iniciarse adecuadamente y controlarse de cerca.Un régimen de tratamiento secuencial (letrozol durante 2 años seguido de tamoxifeno durante 3 años) también podría considerarse como tratamiento adyuvante en función del perfil de seguridad del paciente (ver secciones 4.2, 4.8 y 5.1).
Otras advertencias
Debe evitarse la administración concomitante de Femara con tamoxifeno, otros tratamientos antiestrógenos o que contengan estrógenos, ya que estas sustancias pueden disminuir la acción farmacológica del letrozol (ver sección 4.5).
Como los comprimidos contienen lactosa, Femara no está recomendado para pacientes con problemas hereditarios raros de intolerancia a la galactosa, deficiencia grave de lactasa o malabsorción de glucosa o galactosa.
04.5 Interacciones con otros medicamentos y otras formas de interacción
El metabolismo del letrozol está mediado en parte por CYP2A6 y CYP3A4. La cimetidina, un inhibidor inespecífico débil de las enzimas CYP450, no afectó las concentraciones plasmáticas de letrozol. Se desconoce el efecto de los inhibidores potentes del CYP450.
Hasta la fecha, no existe experiencia clínica sobre el uso de Femara en combinación con estrógenos u otros agentes antineoplásicos, distintos del tamoxifeno. El tamoxifeno, otros antiestrógenos o terapias que contienen estrógenos pueden disminuir la acción farmacológica del letrozol. Además, se ha demostrado que la administración concomitante de tamoxifeno con letrozol reduce sustancialmente las concentraciones plasmáticas de letrozol. Debe evitarse la administración concomitante de letrozol con tamoxifeno, otros agentes antiestrógenos o estrógenos.
In vitro, letrozol inhibe las isoenzimas 2A6 y, moderadamente, 2C19 del citocromo P450, pero se desconoce la relevancia clínica. Por tanto, se debe tener precaución si es necesario administrar letrozol concomitantemente con medicamentos cuya eliminación depende principalmente de estas isoenzimas y cuyo índice terapéutico es estrecho (p. Ej., Fenitoína, clopidrogel).
04.6 Embarazo y lactancia
Mujeres en estado perimenopáusico o en edad fértil
Femara solo debe utilizarse en mujeres con un estado posmenopáusico claramente definido (ver sección 4.4). Dado que hay informes de mujeres que han recuperado la función ovárica durante el tratamiento con Femara a pesar de un claro estado posmenopáusico al inicio del tratamiento, el médico debe analizar la anticoncepción adecuada si es necesario.
El embarazo
Según datos en humanos en los que se han producido casos aislados de defectos congénitos (fusión de labios, genitales ambiguos), Femara puede causar malformaciones congénitas cuando se administra durante el embarazo. Los estudios en animales han demostrado toxicidad reproductiva (ver sección 5.3).
Femara está contraindicado durante el embarazo (ver secciones 4.3 y 5.3).
Hora de la comida
Se desconoce si letrozol / metabolitos se excretan en la leche materna. No se puede excluir un riesgo para los recién nacidos / bebés.
Femara está contraindicado durante la lactancia (ver sección 4.3).
Fertilidad
La acción farmacológica del letrozol es reducir la producción de estrógenos mediante la inhibición de la aromatasa. En mujeres premenopáusicas, la inhibición de la síntesis de estrógenos conduce a aumentos en los niveles de gonadotropinas (LH, FSH). Los niveles elevados de FSH a su vez estimulan el crecimiento folicular y pueden inducir la ovulación.
04.7 Efectos sobre la capacidad para conducir y utilizar máquinas
Femara tiene efectos menores sobre la capacidad para conducir o utilizar máquinas. Se debe tener precaución al conducir o utilizar maquinaria, ya que se han notificado casos de fatiga, mareos y somnolencia poco frecuentes con el uso de Femara.
04.8 Efectos indeseables
Resumen del perfil de seguridad
La frecuencia de las reacciones adversas de Femara se basa principalmente en datos recopilados de estudios clínicos.
Hasta aproximadamente un tercio de los pacientes tratados con Femara en la fase metastásica y aproximadamente el 80% de los pacientes en tratamiento adyuvante, así como en tratamiento adyuvante después del tratamiento estándar con tamoxifeno, experimentaron reacciones adversas. La mayoría de las reacciones adversas se manifestaron durante las primeras semanas de tratamiento. tratamiento.
Las reacciones adversas notificadas con más frecuencia en los ensayos clínicos fueron rubor, hipercolesterolemia, artralgia, fatiga, aumento de la sudoración y náuseas.
Otras reacciones adversas importantes que pueden ocurrir con Femara son: eventos esqueléticos como osteoporosis y / o fracturas óseas y eventos cardiovasculares (incluidos eventos cerebrovasculares y tromboembólicos). La categoría de frecuencia de estas reacciones adversas se describe en la Tabla 1.
Lista tabulada de reacciones adversas
La frecuencia de las reacciones adversas de Femara se basa principalmente en datos recopilados de estudios clínicos.
Las siguientes reacciones adversas, enumeradas en la Tabla 1, se notificaron a partir de estudios clínicos y de la experiencia poscomercialización con Femara:
tabla 1
Las reacciones adversas se clasifican dentro de cada clase de frecuencia, en orden de frecuencia decreciente, utilizando la siguiente convención: muy frecuentes 10%, frecuentes 1% a 10%, poco frecuentes 0,1% a 1%, raras 0,01% a 0,1%, muy raras 0,01% , frecuencia no conocida (la frecuencia no puede estimarse a partir de los datos disponibles).
Infecciones e infestaciones.
Poco frecuentes: infección del tracto urinario.
Neoplasias benignas, malignas y no especificadas (incluidos quistes y pólipos)
Poco frecuentes: dolor tumoral 1
Trastornos del sistema sanguíneo y linfático.
Poco frecuentes: leucopenia.
Trastornos del sistema inmunológico.
Frecuencia no conocida: reacciones anafilácticas.
Trastornos del metabolismo y de la nutrición.
Muy frecuentes: hipercolesterolemia.
Frecuentes: anorexia, aumento del apetito.
Desórdenes psiquiátricos
Frecuentes: depresión.
Poco frecuentes: ansiedad (incluido nerviosismo), irritabilidad
Trastornos del sistema nervioso
Frecuentes: dolor de cabeza, mareos.
Poco frecuentes: somnolencia, insomnio, deterioro de la memoria, disestesia
(incluyendo parestesia, hipoestesia), alteración del gusto, accidente
cerebrovascular, síndrome del túnel carpiano
Trastornos oculares
Poco frecuentes Cataratas, irritación ocular, visión borrosa
Patologias cardiacas
Poco frecuentes: palpitaciones1, taquicardia, episodios isquémicos cardíacos (incluyendo
angina de nueva aparición o agravamiento de la angina, angina de pecho
requiere cirugía, infarto de miocardio e isquemia
miocárdico)
Patologías vasculares
Muy frecuentes: rubor
Frecuentes: hipertensión
Poco frecuentes: tromboflebitis (incluida tromboflebitis venosa superficial y
profundo)
Raras: embolia pulmonar, trombosis arterial, infarto cerebrovascular.
Trastornos respiratorios, torácicos y mediastínicos
Poco frecuentes: disnea, tos
Desórdenes gastrointestinales
Frecuentes: náuseas, dispepsia1, estreñimiento, dolor abdominal, diarrea,
Él vomitó
Poco frecuentes: boca seca, estomatitis 1
Trastornos hepatobiliares
Poco frecuentes: elevación de las enzimas hepáticas.
Frecuencia no conocida: hepatitis.
Trastornos de la piel y del tejido subcutáneo
Muy frecuentes: aumento de la sudoración.
Frecuentes: alopecia, erupción (incluida erupción eritematosa,
maculopapular, similar a la psoriasis y eritema vesicular),
sequedad de la piel
Poco frecuentes: prurito, urticaria.
Frecuencia no conocida: angioedema, necrólisis epidérmica tóxica, eritema multiforme.
Trastornos musculoesqueléticos y del tejido conjuntivo
Muy frecuentes: artralgia.
Común:
Poco común:
Mialgia, dolor óseo1, osteoporosis, fracturas óseas
Artritis
Frecuencia no conocida: chasquido de dedo
Trastornos renales y urinarios.
Poco frecuentes: aumento de la frecuencia urinaria
Enfermedades del aparato reproductor y la mama.
Frecuentes: sangrado vaginal.
Poco frecuentes: secreción vaginal, sequedad vaginal, dolor de mamas.
Desordenes generales y condiciones administrativas del sitio
Muy frecuentes: fatiga (incluyendo astenia, malestar)
Frecuentes: edema periférico.
Poco frecuentes: edema general, membranas mucosas secas, sed, pirexia.
Pruebas de diagnóstico
Frecuentes: aumento de peso.
Poco frecuentes: pérdida de peso.
1 Reacciones adversas a medicamentos notificadas solo en el tratamiento de la fase metastásica
Se han notificado algunas reacciones adversas con diferencias de frecuencia considerables en el tratamiento adyuvante. Las siguientes tablas proporcionan información sobre las diferencias significativas entre Femara versus tamoxifeno solo y entre Femara-tamoxifeno en el tratamiento secuencial:
Tabla 2 Monoterapia adyuvante Femara versus monoterapia con tamoxifeno - Eventos adversos con
Diferencias significativas
Tabla 3 Tratamiento secuencial versus monoterapia con Femara: eventos adversos con diferencias
Significativo
Descripción de reacciones adversas seleccionadas
Reacciones adversas cardiacas
En el tratamiento adyuvante, además de los datos presentados en la Tabla 2, se notificaron las siguientes reacciones adversas para Femara y tamoxifeno, respectivamente (en la mediana de duración del tratamiento de 60 meses más 30 días): angina que requirió cirugía (1,0% frente a 1,0 %); insuficiencia cardíaca (1,1% frente a 0,6%); hipertensión (5,6% frente a 5,7%); accidente cerebrovascular / accidente isquémico transitorio (2,1% frente a 1,9%).
En el tratamiento adyuvante después de la terapia estándar con tamoxifeno, se notificó angina que requirió cirugía (0,8% frente a 0, respectivamente) para Femara (duración media del tratamiento de 5 años) y placebo (duración media del tratamiento 3 años), respectivamente. 6%); angina de nueva aparición o agravación de la angina (1,4% frente a 1,0%); infarto de miocardio (1,0% frente a 0,7%); eventos tromboembólicos * (0,9% frente a 0,3%); ictus / accidente isquémico transitorio * (1,5% frente a 0,8%).
Los eventos marcados con * tuvieron diferencias estadísticamente significativas en los dos grupos de tratamiento.
Reacciones adversas esqueléticas
Para obtener datos de seguridad sobre eventos esqueléticos en el tratamiento adyuvante, consulte la Tabla 2.
En el tratamiento adyuvante después de la terapia estándar con tamoxifeno, significativamente más pacientes tratados con Femara informaron fracturas óseas u osteoporosis (fracturas óseas, 10,4% y osteoporosis 12,2%) que los pacientes del grupo (5,8% y 6, respectivamente, 4%). La mediana de duración del tratamiento fue de 5 años para Femara, en comparación con 3 años para el placebo.
Notificación de sospechas de reacciones adversas
La notificación de sospechas de reacciones adversas que se produzcan tras la autorización del medicamento es importante, ya que permite un seguimiento continuo de la relación beneficio / riesgo del medicamento.Se invita a los profesionales sanitarios a notificar cualquier sospecha de reacciones adversas a través de la Agencia Italiana de Medicamentos. , sitio web: https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse
04.9 Sobredosis
Ha habido informes aislados de sobredosis con Femara.
No se conoce un tratamiento específico para la sobredosis; El tratamiento debe ser sintomático y de soporte.
05.0 PROPIEDADES FARMACOLÓGICAS
05.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: terapias endocrinas. Antagonista hormonal y agentes relacionados: inhibidor de la aromatasa, código ATC: L02BG04.
Efectos farmacodinámicos
"La inhibición de la estimulación del crecimiento celular mediada por estrógenos es un requisito previo para la respuesta tumoral en los casos en que el crecimiento tumoral depende de la presencia de estrógenos y se utiliza terapia endocrina. En mujeres posmenopáusicas, el estrógeno se deriva principalmente de la" acción de la enzima aromatasa, que convierte los estrógenos suprarrenales, principalmente androstenediona y testosterona, en estrona y estradiol, por lo que la supresión de la biosíntesis de estrógenos en los tejidos periféricos y en el propio tejido neoplásico puede conseguirse mediante la inhibición específica de la enzima aromatasa.
El letrozol es un inhibidor de la aromatasa no esteroideo que inhibe la enzima aromatasa al unirse completamente al hemo del citocromo P450, lo que reduce la biosíntesis de estrógenos en todos los tejidos donde está presente.
En mujeres posmenopáusicas sanas, la administración de dosis únicas de 0,1 mg, 0,5 mg y 2,5 mg de letrozol suprime los niveles séricos de estrona y estradiol en un 75% -78% y un 78%, respectivamente, en comparación con los valores iniciales. La supresión máxima se logra en 48-78 horas.
En pacientes posmenopáusicas con cáncer de mama avanzado, las dosis diarias de 0,1-5 mg suprimen las concentraciones plasmáticas de estradiol, estrona y sulfato de estrona en un 75-95% del valor basal en todas las pacientes tratadas. A dosis de 0,5 mg y superiores, muchos valores de estrona y sulfato de estrona están por debajo del umbral de sensibilidad del ensayo; lo que significa que, a estas dosis, se consigue una mayor supresión de la producción de estrógenos. Esta supresión se mantuvo durante la duración del tratamiento en todos los pacientes.
La inhibición de la actividad de la aromatasa por el letrozol es muy específica. No se detectó deterioro de la esteroidogénesis suprarrenal. No se encontraron cambios clínicamente relevantes en las concentraciones plasmáticas de cortisol, aldosterona, 11-desoxicortisol, 17-hidroxi. Progesterona y ACTH, así como en la actividad de la renina plasmática en pacientes posmenopáusicas tratadas con una dosis diaria de 0,1-5 mg de letrozol. La prueba de estimulación con ACTH, realizada a las 6 y 12 semanas de tratamiento con administraciones diarias de 0,1 mg, 0,25 mg, 0,5 mg, 1 mg, 2,5 mg y 5 mg, no indicó ninguna reducción de la producción de aldosterona o cortisol. En consecuencia, no fue necesario administrar
suplementos a base de glucocorticoides y mineralocorticoides.
No se observaron cambios en las concentraciones plasmáticas de andrógenos (androstenediona y testosterona) entre mujeres posmenopáusicas sanas después de dosis únicas de 0.1 mg, 0.5 mg y 2.5 mg de letrozol o en las concentraciones plasmáticas de androstenediona entre pacientes posmenopáusicas tratadas con dosis diarias de
0,1 mg a 5 mg, lo que indica que el bloqueo de la biosíntesis de estrógenos no da como resultado la acumulación de precursores androgénicos. Ni los niveles plasmáticos de LH y FSH ni la función tiroidea, según la evaluación de TSH y la prueba de captación de T3 y T4, se ven afectados por el letrozol.
Tratamiento adyuvante
Estudie BIG 1-98
BIG 1-98 es un estudio multicéntrico, doble ciego, en el que más de 8.000 mujeres posmenopáusicas con cáncer de mama en etapa inicial con receptores hormonales positivos fueron aleatorizadas a uno de los siguientes tratamientos: A. tamoxifeno durante 5 años; B. Femara durante 5 años; C. tamoxifeno durante 2 años seguido de Femara durante
3 años; D. Femara durante 2 años seguido de tamoxifeno durante 3 años.
El criterio de valoración principal fue la supervivencia libre de enfermedad (SSE); los criterios de valoración secundarios de eficacia fueron el tiempo hasta la metástasis a distancia (TDM), la supervivencia libre de enfermedad a distancia (DDFS), la supervivencia general (SG), la supervivencia sin enfermedad sistémica (SDFS), la tasa de cáncer de mama contralateral invasivo y tiempo hasta la recurrencia del cáncer de mama.
Resultados de eficacia con una mediana de seguimiento de 26 y 60 meses
Los datos de la Tabla 4 reflejan los resultados del Análisis de núcleos primarios (PCA) basados en los datos de los grupos de monoterapia.
(A y B) y en los datos de los dos grupos en los que se esperaba el cambio (C y D) a un tratamiento con una mediana de duración de 24 meses y una mediana de seguimiento de 26 meses y a un tratamiento con una mediana de duración de 32 meses y una mediana de seguimiento de 60 meses.
Las tasas de SSE a 5 años fueron del 84% para Femara y del 81,4% para el tamoxifeno.
Tabla 4 Análisis básico principal: supervivencia general y sin enfermedad con una mediana de seguimiento de 26 meses y una mediana de seguimiento de 60 meses (población ITT)
HR = Razón de riesgo; CI = intervalo de confianza
1 Prueba de rangos logarítmicos, estratificada por aleatorización y uso de quimioterapia (sí / no)
2 eventos de SLE: recidiva locorregional, metástasis a distancia, cáncer de mama contralateral invasivo, segunda neoplasia maligna primaria (no mamaria), muerte por cualquier causa sin un evento tumoral previo.
Resultados con una mediana de seguimiento de 96 meses (solo grupos de monoterapia)
El análisis de los grupos de monoterapia (MAA) con actualización a largo plazo de la eficacia de la monoterapia con Femara en comparación con la monoterapia con tamoxifeno (duración media del tratamiento adyuvante: 5 años) se presenta en la Tabla 5.
Tabla 5 Análisis de grupos de monoterapia: supervivencia libre de enfermedad y supervivencia general
con una mediana de seguimiento de 96 meses (población ITT)
1 Prueba de rangos logarítmicos, estratificada por aleatorización y uso de quimioterapia (sí / no)
2 eventos de SSE: recurrencia locorregional, metástasis a distancia, cáncer de mama invasivo
contralateral, segunda neoplasia maligna primaria (no mamaria), muerte por cualquier causa sin un evento de cáncer previo.
3 Observaciones en el grupo de tratamiento con tamoxifeno en el momento del cambio selectivo a letrozol
Análisis de tratamiento secuencial (STA)
El análisis secuencial de tratamiento (STA) aborda la segunda pregunta principal del estudio BIG 1-98, cuyo objetivo es determinar si la secuencia de letrozol y tamoxifeno es superior a la monoterapia con letrozol. No se observaron diferencias significativas en la SSE, SG, SDFS o DDFS entre el cambio y monoterapia (Tabla 6).
Tabla 6 Análisis de tratamientos secuenciales para la supervivencia libre de enfermedad con letrozol como agente endógeno inicial (STA para la población cambiada)
1 Definición del protocolo, incluidas las segundas neoplasias malignas primarias no mamarias después del cambio / durante dos años
2 Ajustado por uso de quimioterapia
No hubo diferencias significativas en DFS, OS, SDFS o DDFS en ninguna de las STA de las comparaciones aleatorias por pares (Tabla 7).
Tabla 7 Análisis de tratamientos secuenciales de aleatorización (STA-R) de supervivencia libre de enfermedad (población ITT STA-R)
1 Ajustado por uso de quimioterapia (sí / no)
2626 (40%) pacientes cambiaron selectivamente a letrozol después de abrir el grupo de tratamiento con tamoxifeno en 2005
Estudio D2407
El estudio D2407 es un estudio de seguridad post-aprobación, abierto, aleatorizado y multicéntrico diseñado para comparar los efectos del tratamiento adyuvante con letrozol y tamoxifeno sobre la densidad mineral ósea (DMO) y los perfiles de lípidos séricos. Un total de 262 pacientes fueron asignados a letrozol durante 5 años o tamoxifeno durante 2 años seguido de letrozol durante 3 años.
A los 24 meses hubo una diferencia estadísticamente significativa en el criterio de valoración principal; la densidad mineral ósea (DMO) en la columna lumbar (L2-L4) mostró una disminución media del 4,1% en el grupo de tratamiento con letrozol en comparación con un aumento medio del 0,3% en el grupo de tratamiento con tamoxifeno.
Ningún paciente con DMO basal normal se volvió osteoporótico durante 2 años de tratamiento y solo 1 paciente con osteopenia basal (puntuación T de -1,9) desarrolló osteoporosis durante el período de tratamiento (evaluación de revisión centralizada).
Los resultados para la DMO de cadera total fueron similares a los observados para la columna lumbar pero menos pronunciados. No hubo diferencias significativas en la tasa de fracturas: 15% en el grupo de tratamiento con letrozol, 17% en el grupo de tratamiento con tamoxifeno.
La mediana de los niveles de colesterol total en el grupo de tratamiento con tamoxifeno disminuyó en un 16% después de 6 meses desde el inicio y esta disminución se mantuvo en las visitas posteriores hasta por 24 meses. En el grupo de tratamiento con letrozol, los niveles de colesterol total se mantuvieron relativamente estables a lo largo del tiempo, mostrando una diferencia estadísticamente significativa a favor del tamoxifeno en cada momento.
Tratamiento adyuvante después de la terapia estándar con tamoxifeno (MA-17)
En un estudio multicéntrico, doble ciego, aleatorizado, controlado con placebo (MA-17) en el que participaron más de 5.100 mujeres posmenopáusicas con cáncer de mama primario desconocido o con receptor positivo que habían completado el tratamiento adyuvante con tamoxifeno (de 4.5 a los 6 años) fueron aleatorizadas a tratamiento con Femara o con placebo durante 5 años.
El criterio de valoración principal fue la supervivencia libre de enfermedad, definida como el intervalo entre la aleatorización y el primer evento de recidiva locorregional, metástasis a distancia o cáncer de mama contralateral.
El primer análisis intermedio programado con una mediana de seguimiento de aproximadamente 28 meses (el 25% de las pacientes fueron seguidas durante al menos 38 meses) mostró que Femara redujo significativamente el riesgo de recurrencia del cáncer de mama en un 42% en comparación con el placebo (HR 0,58). ; IC del 95%: 0,45; 0,76; pag.= 0,00003). El beneficio a favor del letrozol se observó independientemente del estado de los ganglios linfáticos. No hubo diferencias significativas en la supervivencia general: Femara 51 muertes; placebo 62; HR 0,82; IC del 95%: 0,56; 1,19).
En consecuencia, después del primer análisis intermedio, el estudio continuó abierto y, a los pacientes del grupo de tratamiento con placebo se les permitió cambiar a Femara durante 5 años. Más del 60% de los pacientes elegibles (sin enfermedad al inicio del estudio) optaron por cambiarse a Femara. El análisis final incluyó a 1551 mujeres que cambiaron de placebo a Femara en una mediana de 31 meses (rango de 12 meses). A los 106 meses) después de completar el terapia adyuvante con tamoxifeno. La mediana de duración del tratamiento con Femara fue de 40 meses.
Los análisis finales realizados con una mediana de seguimiento de 62 meses confirmaron la reducción significativa en el riesgo de recurrencia del cáncer de mama con Femara.
Tabla 8 Período libre de enfermedad y supervivencia general (población ITT modificada)
HR = Razón de riesgo; CI = intervalo de confianza
1 Cuando se inició el estudio en 2003, 1.551 pacientes en el grupo de tratamiento con placebo aleatorizado (el 60% de ellos eran elegibles para el cambio, es decir, estaban libres de enfermedad) cambiaron al tratamiento con letrozol en una mediana de tiempo de 31 meses después de la aleatorización. Los análisis presentados aquí ignoran el cruce selectivo.
2 Estratificado por estado del receptor, estado de los ganglios linfáticos y quimioterapia adyuvante previa.
3 Definición del protocolo de eventos de supervivencia libre de enfermedad: recidiva locorregional,
metástasis a distancia o cáncer de mama contralateral.
4 Análisis exploratorios de los tiempos de seguimiento en la fecha del cambio (si lo hubiera) en el grupo de tratamiento con placebo.
5 Seguimiento medio de 62 meses.
6 Seguimiento medio hasta la transición (si corresponde) 37 meses.
En el subestudio óseo MA-17 en el que se administraron calcio y vitamina D de forma concomitante, hubo una mayor reducción de la densidad mineral ósea (DMO) desde el inicio con Femara en comparación con el placebo. La única diferencia estadísticamente que se produjo a los 2 años fue en la DMO total de la cadera (disminución mediana con letrozol de 3.8% versus disminución mediana con placebo de
2,0%).
En el subestudio de lípidos MA-17 no hubo diferencias estadísticamente significativas entre letrozol y placebo en el colesterol total o cualquier fracción lipídica.
En el subestudio de calidad de vida actualizado no hubo diferencias significativas entre tratamientos con respecto a la puntuación resumida del componente físico o mental, o en cualquier dominio de puntuación en la escala SF-36. En la escala MENQOL, una mayoría significativa de mujeres en el grupo de tratamiento con Femara en comparación con las que recibieron placebo se sintieron más perturbadas (generalmente en el primer año de tratamiento) por los síntomas resultantes de la privación de estrógenos: rubor y sequedad vaginal. El síntoma más perturbador en la mayoría de los pacientes en ambos grupos de tratamiento fue el dolor muscular, con una diferencia estadísticamente significativa a favor del placebo.
Tratamiento neoadyuvante
Se realizó un estudio doble ciego (P024) en 337 pacientes posmenopáusicas con cáncer de mama aleatorizadas para recibir Femara 2,5 mg durante 4 meses o Tamoxifeno durante 4 meses. Al inicio del estudio, todas las pacientes tenían cánceres positivos en estadio T2-T4c, N0-2, M0, ER y / o PgR y ninguna de las pacientes podía ser elegible para cirugía conservadora de mama. Según la evaluación clínica, se registraron respuestas objetivas en el 55% del grupo de tratamiento con Femara frente al 36% del grupo de tratamiento con tamoxifeno (pag.Ecografía de Femara al 35% versus tamoxifeno al 25%, pag.= 0,04) y de mamografía Femara 34% versus tamoxifeno 16%, pag.P = 0,02) se sometieron a cirugía conservadora de mama. Durante el período de tratamiento preoperatorio de 4 meses, el 12% de los pacientes tratados con Femara y el 17% de los pacientes tratados con tamoxifeno presentaron progresión de la enfermedad en la evaluación clínica.
Tratamiento de primera línea
Se realizó un estudio controlado doble ciego para comparar Femara (letrozol) 2,5 mg y tamoxifeno 20 mg como terapia de primera línea en mujeres posmenopáusicas con cáncer de mama avanzado. En 907 mujeres, el letrozol fue superior al tamoxifeno en cuanto al tiempo de progresión (criterio de valoración principal) y la tasa de respuesta objetiva, el tiempo hasta el fracaso del tratamiento y el beneficio clínico.
Los resultados obtenidos se resumen en la Tabla 9:
Tabla 9 Resultados con una mediana de seguimiento de 32 meses
El tiempo hasta la progresión fue significativamente más largo y la tasa de respuesta significativamente más alta para letrozol independientemente de si se administró o no terapia adyuvante antiestrógeno. El tiempo hasta la progresión fue significativamente más largo para el letrozol independientemente del sitio dominante de la enfermedad. La mediana del tiempo hasta la progresión fue de 12,1 meses para Femara y de 6,4 meses para el tamoxifeno en pacientes con el sitio de la enfermedad solo en tejidos blandos y una mediana de 8,3 meses para Femara y 4,6 meses para el tamoxifeno en pacientes con metástasis viscerales.
El diseño del estudio permitió a los pacientes pasar a una terapia alternativa o interrumpir el estudio al progresar la enfermedad.Aproximadamente el 50% de los pacientes pasaron al grupo de tratamiento opuesto y, de hecho, el cruce se completó en 36 meses. La mediana del tiempo de transición fue de 17 meses (Femara a tamoxifeno) y 13 meses (tamoxifeno a Femara).
El tratamiento de primera línea del cáncer de mama avanzado dio como resultado una mediana de supervivencia general para Femara de 34 meses frente a 30 meses para el tamoxifeno (prueba de rango logarítmico P = 0,53, no significativo). La falta de una ventaja para Femara en la supervivencia general puede explicarse por el diseño cruzado del estudio.
Tratamiento de segunda línea
En mujeres posmenopáusicas con cáncer de mama avanzado, previamente tratadas con antiestrógenos, se realizaron dos ensayos clínicos bien controlados comparando dos dosis de letrozol (0,5 mg y 2,5 mg) y megestrol, respectivamente, acetato y aminoglutetimida.
El tiempo de progresión no fue significativamente diferente entre 2,5 mg de letrozol y acetato de megestrol (pag.= 0,07). Hubo diferencias estadísticamente significativas a favor de letrozol 2,5 mg versus acetato de megestrol con respecto a la tasa de respuesta tumoral objetiva global (24% versus 16%, pag.= 0.04), y el tiempo hasta el fracaso del tratamiento (pag.= 0,04). La supervivencia global no fue significativamente diferente entre los 2 grupos (pag.=0,2).
En el segundo estudio, la tasa de respuesta no fue significativamente diferente entre letrozol 2,5 mg y aminoglutetimida (pag.= 0,06). El letrozol 2,5 mg fue estadísticamente superior a la aminoglutetimida en el tiempo hasta la progresión (pag.= 0,008), tiempo hasta el fracaso del tratamiento (pag.= 0,003) y supervivencia global (pag.=0,002).
Cáncer de mama masculino
No se ha estudiado el uso de Femara en hombres con cáncer de mama.
05.2 Propiedades farmacocinéticas
Absorción
El letrozol se absorbe rápida y completamente en el tracto gastrointestinal (biodisponibilidad absoluta media: 99,9%). Los alimentos reducen ligeramente la tasa de absorción (mediana de Tmax 1 hora en ayunas frente a 2 horas después de las comidas; y media de Cmax 129 ± 20,3 nmol / litro en ayunas frente a 98,7 ± 18,6 nmol / litro después de las comidas) pero el grado de absorción (AUC) sí lo hace. no variar. Se considera que este modesto efecto sobre la velocidad de absorción no tiene relevancia clínica y, por lo tanto, letrozol puede tomarse con o sin comidas.
Distribución
La unión de letrozol a las proteínas plasmáticas es aproximadamente del 60%, de las cuales la mayor parte (55%) se une a la albúmina. La concentración de letrozol en los eritrocitos es aproximadamente el 80% del nivel plasmático. Después de la administración de 2,5 mg de letrozol marcado con 14C, aproximadamente el 82% de la radiactividad plasmática es el compuesto original La exposición sistémica a los metabolitos es baja. El letrozol se distribuye rápida y ampliamente en los tejidos. Su volumen aparente de distribución en estado estacionario es de aproximadamente 1,87 ± 0,47 L / kg.
Biotransformación
La principal vía de eliminación de letrozol está representada por el aclaramiento metabólico con la formación de un metabolito farmacológicamente inactivo, carbinol CLm = 2,1 l / h, pero es relativamente lento en comparación con el flujo sanguíneo hepático (aproximadamente 90 l / h). Las isoenzimas 3A4 y 2A6 del citocromo P450 son capaces de convertir letrozol en este metabolito. La formación de estos metabolitos menores no identificados y la excreción renal y fecal directa juegan un papel menor en la eliminación general de letrozol. Después de la administración de 2.5 mg de letrozol marcado con 14C a voluntarios sanos en la posmenopausia, el 88.2 ± 7.6% de la radioactividad se recuperó en el orina y 3,8 ± 0,9% en heces en 2 semanas. Al menos el 75% de la radiactividad recuperada en la orina hasta 216 horas (84,7 ± 7,8% de la dosis) se atribuyó al glucurónido del metabolito carbinol, aproximadamente el 9% a dos metabolitos no identificados y el 6% al letrozol inalterado.
Eliminación
La vida media de eliminación terminal aparente es de aproximadamente 2 a 4 días. Después de la administración diaria de 2,5 mg, se alcanzó el estado de equilibrio en 2-6 semanas. Las concentraciones plasmáticas en el estado de equilibrio son aproximadamente 7 veces más altas que las concentraciones detectadas después de una dosis única de 2,5 mg, mientras que son 1,5 a 2 veces más altos que los valores esperados en el estado estacionario basados en las concentraciones detectadas después de una dosis única, esto sugiere que existe una ligera falta de linealidad de la farmacocinética de letrozol después de la administración diaria. administración de 2,5 mg Dado que los niveles en estado estacionario se mantienen a lo largo del tiempo, se puede concluir que no hay una acumulación continua de letrozol.
Linealidad / no linealidad
La farmacocinética de letrozol fue proporcional a la dosis después de dosis orales únicas de hasta 10 mg (rango de dosis: 0,01-30 mg) y después de dosis diarias de hasta 1,0 mg (rango de dosis: 0,1 - 5 mg). Después de una dosis oral única de 30 mg, se produjo un pequeño aumento proporcional a la dosis en el valor de AUC. Es probable que la sobreproporcionalidad sea el resultado de una saturación de los procesos metabólicos de eliminación. Se alcanzaron niveles estables después de 1-2 meses en todos los regímenes de dosificación probados (0,1-5,0 mg por día).
Poblaciones especiales
Pacientes de edad avanzada
La edad no tiene ningún efecto sobre la farmacocinética del letrozol.
Insuficiencia renal
En un estudio en el que participaron 19 voluntarios con diversos grados de función renal (aclaramiento de creatinina de 24 horas de 9-116 ml / min), no se encontró ningún efecto sobre la farmacocinética del letrozol después de una dosis única de 2,5 mg. Además de este estudio que evalúa la influencia de la insuficiencia renal sobre el letrozol, se realizó un análisis de covariables sobre los datos de dos estudios fundamentales (Estudio AR / BC2 y Estudio AR / BC3).
Aclaramiento de creatinina calculado (CLcr) [Estudio AR / BC2: rango: 19 - 187 ml / min; Estudio AR / BC3: rango: 10-180 ml / min] no demostró una asociación estadísticamente significativa entre los niveles mínimos de letrozol en plasma en estado estacionario (Cmin). Además, los datos del estudio AR / BC2 y del estudio AR / BC3 en el cáncer de mama metastásico de segunda línea no mostraron evidencia de un efecto negativo del letrozol sobre el CLcr o de la función renal alterada.
Por tanto, no es necesario ajustar la dosis en pacientes con insuficiencia renal (CLcr ≥ 10 ml / min). Se dispone de poca información en pacientes con insuficiencia renal grave (CLcr
Deterioro hepático
En un estudio similar en sujetos con diversos grados de función hepática, los valores medios del AUC en voluntarios con insuficiencia hepática moderada (clase B de Child-Pugh) fueron un 37% más altos que en sujetos normales, pero aún dentro de los límites observados en sujetos sin función hepática alterada. La farmacocinética del letrozol se evaluó en un estudio comparativo en el que, tras la administración de una dosis oral única en ocho sujetos varones con cirrosis hepática e insuficiencia hepática grave (Child-Pugh clase C) y en voluntarios sanos (N = 8), el área bajo la curva AUC y la vida media t½ aumentaron en un 95 y un 187%, respectivamente. Por tanto, Femara debe administrarse a estos pacientes con precaución y tras una cuidadosa consideración de la posible relación riesgo / beneficio.
05.3 Datos preclínicos sobre seguridad
No hubo evidencia de toxicidad sistémica o de órganos diana en una serie de estudios de toxicología preclínicos realizados con especies animales estándar.
La toxicidad aguda del letrozol fue baja en roedores expuestos a dosis de hasta 2000 mg / kg. En perros, letrozol indujo signos de toxicidad moderada a dosis de hasta 100 mg / kg.
En el contexto de estudios toxicológicos para administración repetida en ratas y perros, con una duración de hasta 12 meses, los principales resultados observados pueden atribuirse a la actividad farmacológica del compuesto. La dosis sin eventos adversos fue de 0,3 mg / kg en ambas especies.
La administración oral de letrozol a ratas hembra dio como resultado una reducción en la proporción de apareamiento-preñez y un aumento de las pérdidas antes de la implantación.
Los estudios sobre el potencial mutagénico del letrozol realizados tanto in vitro ese en vivo no documentó ninguna evidencia de genotoxicidad.
En un estudio de carcinogenicidad de 104 semanas en ratas macho, no se detectaron tumores relacionados con el tratamiento. En ratas hembras, se encontró una reducción en la incidencia de tumores mamarios tanto benignos como malignos con todas las dosis de letrozol utilizadas.
En un estudio de carcinogenicidad en ratones de 104 semanas, no se detectaron tumores relacionados con el tratamiento en ratones machos. En ratones hembra, se observó un aumento generalmente relacionado con la dosis en la incidencia de tumores benignos de las células ováricas de la teca granulosa con todas las dosis de letrozol probadas. Estos tumores se consideraron relacionados con la inhibición farmacológica de la síntesis de estrógenos y pueden ser causados por una aumento de la LH como resultado de una disminución de los estrógenos circulantes.
En ratas y conejas preñadas, se demostró que letrozol es embriotóxico y fetotóxico tras la administración oral a dosis clínicamente relevantes. En ratas que dieron a luz a fetos vivos, hubo una mayor incidencia de malformaciones fetales, incluida la cabeza abovedada y la fusión vertebral cervical / central. No se observó un aumento de malformaciones fetales en conejos. Se desconoce si estas malformaciones fueron una consecuencia indirecta de las propiedades farmacológicas (inhibición de la biosíntesis de estrógenos) o un efecto directo del fármaco (ver secciones 4.3 y 4.6).
Las observaciones que surgen de los estudios preclínicos se limitan a las asociadas a la actividad farmacológica conocida, que representa la única área de preocupación en términos de seguridad para su uso en humanos derivada de la extrapolación de estudios realizados en animales.
06.0 INFORMACIÓN FARMACÉUTICA
06.1 Excipientes
Contenido del comprimido: lactosa monohidrato, celulosa microcristalina, almidón de maíz, carboximetil almidón de sodio, estearato de magnesio y sílice coloidal anhidra.
Recubrimiento: hipromelosa, talco, macrogol 8000, dióxido de titanio (E171) y óxido de hierro amarillo (E172).
06.2 Incompatibilidad
Irrelevante
06.3 Período de validez
5 años
06.4 Precauciones especiales de conservación
No conservar por encima de 30 ° C.
Almacene en el paquete original para proteger el medicamento de la humedad.
06.5 Naturaleza del envase primario y contenido del envase.
Blísteres de PVC / PE / PVDC / aluminio.
Envases de 10 (1 x 10), 14 (1 x 14), 28 (2 x 14), 30 (3 x 10), 100 (10 x 10) comprimidos
Es posible que no se comercialicen todos los tamaños de envases.
06.6 Instrucciones de uso y manipulación
Sin instrucciones especiales
07.0 TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Novartis Europharm Limited
Carretera de Wimblehurst
Horsham
West Sussex, RH12 5AB Reino Unido
08.0 NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
30 tabletas: 033242013
100 tabletas: 033242025
10 comprimidos 033242037
14 comprimidos 033242049
28 tabletas 033242052
09.0 FECHA DE LA PRIMERA AUTORIZACIÓN O RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 21.03.1997
Fecha de renovación: 24.07.2006