Una condición hereditaria, la AME se debe a mutaciones en el gen SMN1 o en el gen SMN2, cuyo propósito es producir una proteína que sirve para asegurar la supervivencia de las neuronas motoras.
Hay cinco formas diferentes de atrofia muscular espinal: tipo 0, tipo 1, tipo 2, tipo 3 y tipo 4. Los tres primeros tipos son muy graves y provocan la muerte prematura del paciente; el tipo 3 y el tipo 4 son variantes más leves, que afectan el nivel de vida del paciente, pero sin provocar una muerte prematura.
Se necesita una prueba genética en una muestra de sangre para diagnosticar la AME.
Actualmente, la terapia de la AME se basa principalmente en tratamientos sintomáticos, dirigidos a aliviar los trastornos y controlar las complicaciones. Existe una cura disponible, basada en los principios de la terapia génica, pero es una solución muy cara y aplicable solo a ciertos pacientes.
, que se manifiesta con atrofia y consecuente debilitamiento de la musculatura esquelética y dificultades motoras.
La AME es una condición que puede causar la muerte del paciente a una edad temprana o muy temprana: las formas más graves de la enfermedad, de hecho, afectan la eficiencia de los músculos respiratorios y son responsables de episodios de insuficiencia respiratoria o neumonía con un desenlace fatal.
Neuronas motoras y SMA

Las motoneuronas, o motoneuronas, son células nerviosas que surgen en el sistema nervioso central (cerebro y médula espinal) y que, mediante sus extensiones (axones), controlan la actividad de músculos y glándulas.
Hay dos tipos de motoneuronas: motoneuronas superiores (o primeras motoneuronas) y motoneuronas inferiores (o segundas motoneuronas).
Las motoneuronas superiores se originan en el cerebro y dirigen la actividad de las motoneuronas inferiores, que surgen principalmente en la médula espinal y son responsables de dirigir la actividad de los músculos esqueléticos (o somáticos), de los músculos lisos (o viscerales), del músculo cardíaco y del corazón.
Las neuronas motoras de los individuos con AME se degeneran gradualmente, provocando "atrofia muscular por inactividad que, en los casos más graves, resulta en parálisis, insuficiencia respiratoria y muerte".
Epidemiología: ¿que tan común es la atrofia muscular espinal?
La AME tiene una "incidencia anual de 1 caso por cada 10,000 nuevos nacimientos.
5 y de la que depende la producción de la denominada proteína de supervivencia de las neuronas motoras (SMN).Como sugiere el nombre de la proteína producida por SMN1 y SMN2, la mutación de estos genes priva a las neuronas motoras de una sustancia biológica esencial para su supervivencia; más precisamente, reduce los niveles de proteína: por ejemplo, en presencia de mutaciones en SMN1, los niveles de proteína SMN caen al 10-20% de lo normal.
Evidentemente, la ausencia de cantidades adecuadas de la proteína SMN determina la degeneración progresiva de las neuronas motoras.
La pérdida de motoneuronas interrumpe la señalización nerviosa que permite controlar la actividad de los músculos del cuerpo humano; estos últimos, como consecuencia de que ya no son utilizables, sufren un proceso paulatino de atrofia y debilitamiento.
Sabía usted que ...
El gen SMN2 es, para SMA, un gen modificador de la enfermedad; de hecho, en pacientes con mutación en SMN1 y que tienen, por alguna razón, tres o cuatro copias del gen SMN2, la AME se presenta en una forma más leve.
Atrofia muscular espinal: tipos de mutación
Cuando la AME se debe a una "alteración de SMN1, en el 95-98% de los casos la mutación responsable consiste en una deleción del gen completo, mientras que sólo en el 2-5% en una" anomalía de la secuencia normal del gen.
Atrofia muscular espinal: una enfermedad hereditaria
En casi todos los casos (98%), la anomalía genética responsable de la AME es hereditaria, es decir, son los padres del enfermo quienes la transmiten.
El 2% de los casos no hereditarios de AME se deben a una mutación. de novo Ocurrió en una etapa muy temprana del desarrollo embrionario.
Modelo de SMA y herencia
El modelo de herencia para la atrofia muscular espinal es autosómico recesivo, lo que significa que, para heredar la AME, es fundamental que ambos padres sean portadores sanos del defecto genético en SMN1 o SMN2 y que ambos padres lo transmitan.
En el caso de enfermedades hereditarias autosómicas recesivas como la AME, la probabilidad de que ambos portadores sanos transmitan el defecto genético al niño, enfermándolo, es del 25%, o uno de cada 4 casos.
Tipos de AME
Según la edad de aparición y la gravedad de la afección, los expertos reconocen cinco formas diferentes de atrofia muscular espinal:
- SMA tipo 0: es la forma más grave de todas. Se manifiesta incluso antes del nacimiento con movilidad reducida del feto.
Los bebés generalmente sobreviven unas semanas después del nacimiento, incluso cuando reciben asistencia respiratoria. - AME tipo 1: de las formas que ocurren durante la vida, es la más grave y común (alrededor del 50% de los casos); aparece a una edad temprana, generalmente dentro del sexto mes de vida.
Por regla general, es causa de muerte ya en los primeros años de vida; raramente, durante la adolescencia.
La muerte ocurre típicamente por "insuficiencia respiratoria o" infección pulmonar. - SMA tipo 2: es la forma que, por gravedad, ocupa el segundo lugar; generalmente, comienza entre los 7 y los 18 meses de vida.
La esperanza de vida de los afectados es mayor que en el caso anterior: los pacientes, de hecho, logran llegar a la edad adulta. - AME tipo 3: menos grave que las dos anteriores, esta forma de AME suele aparecer después de los 18 meses de vida (en algunos casos, también puede aparecer durante la niñez o la adolescencia).
Implica discapacidades importantes, pero no afecta la esperanza de vida. - AME tipo 4: es la forma adulta de la enfermedad y la menos grave; generalmente comienza alrededor de la tercera década de la vida y tiene un curso muy lento.
Por lo general, no es responsable de los problemas respiratorios y está asociado con una "esperanza de vida normal".
Los niveles de proteína SMN afectan la gravedad de la AME: cuanto menor es la cantidad de SMN, mayor es la gravedad de la enfermedad relacionada.
La reducción en los niveles de SMN está estrechamente relacionada con la extensión del defecto genético que afectó a los genes SMN1 o SMN2: cuanto más extenso es este defecto, más significativa es la reducción en la cantidad de proteína SMN (este es el caso, por ejemplo, de una deleción de un gen).
Además, la AME no compromete las funciones intelectuales (el coeficiente intelectual de los pacientes es normal) y preserva el órgano de la vista.
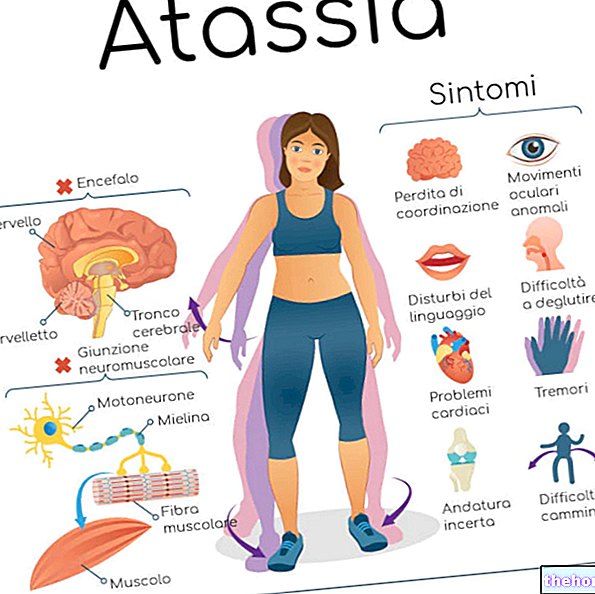
Para más información: AME: todos los síntomasSíntomas de AME tipo 0
Como se indicó anteriormente, la AME de tipo 0 ya se presenta en la edad prenatal con movilidad fetal reducida; al nacer, entonces, el niño enfermo presenta evidentes dificultades para tragar y respirar.
La enfermedad provoca la muerte a las pocas semanas del nacimiento, incluso cuando el paciente recibe asistencia respiratoria.
Síntomas de AME tipo 1
Los niños con AME tipo 1 tienen músculos muy débiles que no se desarrollan como deberían (atrofia muscular). Esto les impide realizar actividades como levantar la cabeza, mover las extremidades y adoptar una posición sentada; además, complica progresivamente funciones vitales, como succionar la leche, tragar, masticar y respirar.
Por lo general, la AME tipo 1 es fatal durante los primeros años de vida; algunos pacientes, sin embargo, logran llegar a la adolescencia.
La muerte generalmente ocurre por insuficiencia respiratoria o una "infección pulmonar debido a dificultades para tragar (neumonía por ingestión o neumonía ab ingestis).
Síntomas de AME tipo 2
SMA tipo 2 se manifiesta clásicamente con:
- Suavidad de los músculos de brazos y piernas;
- Temblores en los dedos y las manos;
- Dificultad para asumir la posición sentada de forma independiente (el paciente, sin embargo, logra mantenerla);
- Dificultad para pararse y caminar.
- Deformidad y problemas articulares;
- Dificultad para respirar y tragar alimentos;
- Escoliosis (suele aparecer más tarde).
Incluso en esta situación, las dificultades respiratorias y la ingestión de alimentos son la causa de la muerte prematura, que generalmente ocurre al comienzo de la edad adulta.
Síntomas de AME tipo 3
La AME tipo 3 causa problemas con la postura y el equilibrio, temblores de manos y dificultad para levantarse de una posición sentada, caminar, subir escaleras y correr.
Al principio, las dolencias no requieren apoyo para la locomoción; posteriormente, con la degeneración de un mayor número de motoneuronas, las muletas, andadores y sillas de ruedas se vuelven fundamentales.
Aunque puede suceder, es muy raro que los pacientes con AME tipo 3 sufran problemas respiratorios y traguen alimentos.
En presencia de esta forma de AME, la esperanza de vida es normal, pero con todos los problemas antes mencionados.
Síntomas de AME tipo 4
Con el inicio en la edad adulta, la AME tipo 4 se asocia típicamente con:
- Debilitamiento del tono muscular en brazos y piernas;
- Dificultad para caminar
- Temblores y contracciones repentinas de los músculos.
Inicialmente, las quejas mencionadas son moderadas; en la vejez, se vuelven más consistentes.
Al igual que la AME de tipo 3, la AME de tipo 4 no es una enfermedad que afecte la esperanza de vida del paciente.
AME: ¿cuando ver a un médico?
Se recomienda encarecidamente a todos los padres que sepan que son portadores sanos de AME que consulten a un pediatra con experiencia en enfermedades genéticas y a un genetista.
Si no tienes información de este tipo, es bueno evaluar mes a mes el desarrollo motor de tu hijo y las funciones de las que depende la vida (ej .: respiración).
Ciertamente, la incapacidad para sentarse o asumir la posición sentada, la dificultad para alimentarse, la presencia de déficits respiratorios y una musculatura delgada y menos tonificada que la de los compañeros constituyen señales de alarma.
En cuanto a la forma adulta de AME, se sospecha y debe controlarse la aparición más o menos repentina de debilidad muscular y dificultad para caminar.
Atrofia muscular espinal: complicaciones
Las formas más graves de AME pueden provocar complicaciones como:
- Asfixia por comida. Se debe a la capacidad reducida para masticar e ingerir alimentos.
- Insuficiencia respiratoria. Es consecuencia de la incapacidad para controlar la actividad de los músculos respiratorios.
- Neumonía ab ingestis (o neumonía por inhalación). Ocurre cuando un material extraño que transporta patógenos, como alimentos, saliva o secreciones nasales, ingresa o se acumula en los pulmones.
Neumonía ab ingestis es el resultado de dificultades para tragar. - Parálisis que resulta en el uso de sillas de ruedas. Ocurre cuando la enfermedad ha comprometido irremediablemente las facultades locomotoras del paciente.
- Desnutrición. Es otra consecuencia de la dificultad para tragar: el paciente, de hecho, lucha por alimentarse adecuadamente.
Cabe señalar que, a veces, se pueden utilizar pruebas como la electromiografía o la biopsia muscular durante el diagnóstico de AME.
SMA: examen físico y anamnesis
El examen físico en un paciente que puede sufrir AME implica un análisis cuidadoso de los síntomas y la búsqueda de algunos signos típicos de la enfermedad, como:
- Debilidad y ternura de los músculos;
- Contracciones musculares repentinas.
- Reflejos tendinosos reducidos o ausentes.
En cuanto a la historia clínica, sin embargo, esta se centra principalmente en la historia familiar del paciente, con el fin de establecer si algún otro miembro de la familia (padres, hermanos, abuelos) se queja o se queja de una sintomatología similar. Obviamente, el hecho de que la AME sea hereditaria enfermedad, transmitida de los padres.
Si bien no permiten realizar un diagnóstico definitivo, la exploración física y la historia clínica pueden aportar información de gran utilidad, que orienta las investigaciones hacia la realización de una prueba genética.
Claramente, si el paciente es un niño pequeño, los padres interactuarán con el médico durante el historial médico.
SMA y prueba genética

La prueba genética para la detección de AME implica la búsqueda y estudio de mutaciones en los genes SMN1 / SMN2 en una muestra de células sanguíneas del paciente.
La presencia de alteraciones genéticas obviamente significa enfermedad.
El análisis de las mutaciones detectadas es fundamental para establecer el tipo de atrofia muscular espinal presente y la gravedad de la afección.
Para conocer los resultados de la mencionada prueba genética, generalmente es necesario esperar de 3 a 4 semanas (los tiempos de espera precisos varían según el centro genético que realiza la prueba).
AME: ¿es posible el diagnóstico prenatal?
Es posible diagnosticar AME en la edad prenatal.
Para hacer esto, necesita una prueba genética en una muestra de células fetales, obtenida a través de métodos delicados como la villocentesis o la amniocentesis.
Dado el riesgo de aborto que caracteriza a la CVS y la amniocentesis, los médicos realizan investigaciones prenatales para detectar cualquier mutación atribuible a la "atrofia muscular espinal sólo si hay antecedentes familiares de AME o si el feto es hijo de portadores sanos de la enfermedad".
SMA y detección neonatal
Cabe señalar que en un par de regiones italianas (Lazio y Toscana) hay un servicio activo poner en pantalla para el diagnóstico temprano de AME y otras enfermedades genéticas graves.
El diagnóstico precoz de estas enfermedades permite planificar oportunamente la terapia sintomática más adecuada para el control de síntomas y complicaciones.
Atrofia muscular espinal y planificación de un embarazo
Se recomienda la asesoría genética para todas las mujeres que buscan un embarazo y que:
- Tuvieron un hijo con AME en un embarazo anterior;
- Tienen antecedentes familiares de AME detrás de ellos;
- ¿Son portadores sanos de la enfermedad o su pareja lo es?
La asesoría genética puede ayudar a las mujeres con estas afecciones a comprender los riesgos a los que está expuesto un futuro hijo.
SMA y diagnóstico diferencial
Hay dos patologías muy similares a la AME, que solo una "investigación diagnóstica exhaustiva reconoce y previene la confusión con la" atrofia muscular espinal: estas son la "atrofia muscular espinal con dificultad respiratoria (SMARD) y la" atrofia muscular bulbo-espinal (BSMA). O Enfermedad de Kennedy); el primero se debe a una mutación del gen IGHMBP2 ubicado en el cromosoma 11, mientras que el segundo se debe a una mutación del cromosoma X sexual.
y productos farmacéuticos) aprobó Zolgensma, el método de terapia génica para el tratamiento de la atrofia muscular espinal.
Zolgensma consiste en una técnica de biología molecular muy avanzada, que incluye el uso de un virus-vector capaz de insertar una copia normal del gen SMN1 / SMN2 en el ADN presente dentro de las neuronas motoras de un paciente.
La administración del virus-vector antes mencionado se realiza mediante inyección intravenosa.
Zolgensma resultó eficaz. Sin embargo, como se anticipó, tiene dos límites principales que impiden su uso común:
- Es muy caro. Se habla de millones de euros;
- Solo es aplicable a pacientes con AME menores de 2 años.
Atrofia muscular espinal: tratamientos sintomáticos
Las terapias sintomáticas para la AME garantizan mayores beneficios si se adoptan con prontitud; esto hace que el diagnóstico precoz de la enfermedad sea muy importante.
SMA y soporte respiratorio
El apoyo respiratorio adecuado ayuda a los que sufren de AME no solo a respirar, sino también a reducir el riesgo de infecciones pulmonares.
Entre las diversas opciones terapéuticas, se encuentran las mascarillas para ventilación no invasiva y soluciones más invasivas como la intubación orotraqueal y la traqueotomía; las primeras son ideales para casos menos severos, mientras que las soluciones más invasivas son fundamentales para pacientes con problemas respiratorios graves.
SMA y apoyo nutricional
Las formas más graves de atrofia muscular espinal afectan la capacidad de tragar y masticar alimentos, exponiendo al paciente al riesgo de asfixia, neumonía por ingestión y desnutrición.
Para controlar estas peligrosas consecuencias, es fundamental recurrir a ayudas para la alimentación, como la sonda nasogástrica o la cirugía de gastrostomía, y contar con un nutricionista que planifique una dieta adecuada a las necesidades del paciente.
AME y fisioterapia
Las dificultades motoras que caracterizan al paciente con atrofia muscular espinal provocan rigidez articular y muscular por inactividad.
Un programa de fisioterapia adecuado permite mejorar, en la medida de lo posible, la flexibilidad de los músculos y hacer que las articulaciones sean menos rígidas.
Claramente, este programa incluye ejercicios cuya ejecución está al alcance de las capacidades del paciente.
AME y ortopedia
En presencia de escoliosis, típica de las formas graves de AME, es fundamental consultar a un ortopedista; esto último podría indicar el uso de un corsé ortopédico, si la deformación es leve, u optar por la cirugía de fusión espinal, si la malformación espinal es severa.
Medicamentos contra la AME
Desde hace algunos años también existen fármacos específicos contra la AME.
Estos medicamentos merecen un tratamiento aparte en comparación con las terapias sintomáticas, aunque no permiten curar la enfermedad, sino solo contenerla.
Los fármacos específicos contra la AME actualmente disponibles son Spinraza (nusinersen) y Evrysdi (risdiplam): el primero actúa corrigiendo la producción aberrante de la proteína SMN en el proceso; el segundo aumenta los niveles de producción de SMN, tratando también de mantenerlos en un nivel cuota adecuada a las necesidades del organismo humano.
Aprobados por la FDA en 2017 y 2020 respectivamente, Spinraza y Evrysdi garantizan resultados, en algunos casos incluso más que satisfactorios, sin embargo tienen una limitación importante: son muy costosos.
Para más información: Spinraza: cómo funciona, riesgos y beneficios



.jpg)










.jpg)