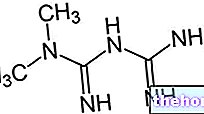
Ingredientes activos: pravastatina (sal sódica de pravastatina)
APLACTIN 20 mg comprimidos
APLACTIN 40 mg comprimidos
¿Por qué se usa Aplactin? ¿Para qué sirve?
APLACTIN contiene pravastatina, sustancia perteneciente al grupo de las estatinas, fármacos capaces de reducir el nivel de grasas y colesterol en sangre. Esta acción es posible gracias al bloqueo de una enzima importante en la síntesis del colesterol en sí, llamada HMG-CoA reductasa.
APLACTIN está indicado en adultos en los siguientes casos:
- Hipercolesterolemia (exceso de colesterol en la sangre)
Tratamiento de la hipercolesterolemia primaria (familiar) o dislipidemia mixta (cambios en la cantidad de grasa en la sangre) como complemento de la dieta, cuando la respuesta a la dieta u otros tratamientos no farmacológicos (por ejemplo, ejercicio o reducción de peso) ha sido inadecuada.
- Prevención primaria
Reducción de la mortalidad y la frecuencia de enfermedades cardiovasculares (que afectan al corazón y / o vasos sanguíneos) en pacientes con hipercolesterolemia moderada a grave y con alto riesgo de un primer evento cardiovascular (infarto, ictus), además de la dieta.
- Prevención secundaria
Reducción de la mortalidad y la frecuencia de enfermedades cardiovasculares en pacientes que han tenido antecedentes de infarto de miocardio (muerte de parte del tejido cardíaco causada por el cierre de un vaso que lleva sangre al corazón) o angina de pecho inestable (dolor en el pecho). debido a una disminución temporal del flujo sanguíneo y del oxígeno al corazón) y que tengan niveles de colesterol normales o elevados, además de corregir otros factores de riesgo.
- Postrasplante
Reducción de la hiperlipidemia (aumento de los niveles de grasa en sangre) tras el trasplante en pacientes sometidos a terapia inmunosupresora (terapia para evitar el rechazo del órgano trasplantado) tras el trasplante de un órgano sólido (p. Ej., Hígado, páncreas, riñones) (ver sección 3 "Cómo tomar APLACTIN "y" Otros medicamentos y APLACTIN ").
Contraindicaciones Cuando no se debe usar Aplactin
No tome APLACTIN
- Si es alérgico a la pravastatina oa cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- Si tiene una enfermedad hepática activa (enfermedad hepática) y si tiene un aumento marcado y duradero de los niveles de transaminasas en sangre (enzimas que indican la función hepática), que superan el triple de los límites máximos permitidos (consulte "Advertencias y precauciones"). .
- Si está embarazada o amamantando (consulte "Embarazo y lactancia").
Precauciones de uso Lo que necesita saber antes de tomar Aplactin
Hable con su médico o farmacéutico antes de tomar APLACTIN.
Antes de iniciar el tratamiento con APLACTIN, informe a su médico:
- si padece insuficiencia renal (función renal alterada);
- si tiene hipotiroidismo (disminución de la función tiroidea);
- si anteriormente ha tenido problemas de toxicidad muscular por 'usar estatinas y fibratos (ver' Otros medicamentos y APLACTIN ') u otro medicamento para reducir el colesterol, como el ácido nicotínico (niacina);
- si usted o alguien de su familia tiene trastornos musculares hereditarios;
- si padece alcoholismo (dependencia del alcohol);
- si tiene más de 70 años, especialmente si tiene otros factores que podrían predisponerle a trastornos musculares;
- si está tomando o ha tomado en los últimos 7 días un medicamento llamado ácido fusídico (utilizado para tratar infecciones bacterianas) por vía oral o por inyección; la combinación de ácido fusídico con APLACTIN puede causar problemas musculares graves (rabdomiólisis).
En estos casos, el médico deberá realizar un análisis de sangre para evaluar los niveles de creatina quinasa (CK) antes de comenzar la terapia.
Informe a su médico de inmediato si experimenta síntomas como dolor, tensión, debilidad o calambres musculares de naturaleza desconocida durante el tratamiento, porque se necesitan análisis de sangre para evaluar los niveles de creatina quinasa (CK), o puede ser necesario dejar de tomar el tratamiento. Este medicamento puede causar problemas musculares como mialgia (dolor muscular), miopatía (enfermedad que afecta a los músculos) Rabdomiólisis (enfermedad caracterizada por lesión de las fibras musculares), incluso mortal, puede ocurrir muy raramente, que puede estar asociada con insuficiencia renal secundaria (alteración de la función renal resultante de otras patologías).
Además, informe a su médico o farmacéutico si tiene debilidad muscular constante. Es posible que se necesiten pruebas y medicamentos adicionales para diagnosticar y tratar esta afección.
Deje de tomar este medicamento y consulte a su médico si experimenta alguno de los siguientes síntomas, especialmente si está siendo tratado con este medicamento durante un período prolongado, ya que puede producirse una enfermedad pulmonar intersticial (problema pulmonar):
- disnea (dificultad para respirar);
- tos seca;
- cansancio;
- pérdida de peso y fiebre.
Mientras esté en tratamiento con este medicamento, su médico comprobará los niveles de enzimas producidas por el hígado (AST y ALT) y le indicará que deje de tomar APLACTIN si los niveles son tres veces superiores a lo normal y de forma constante.
Informe a su médico si ha padecido una enfermedad hepática (enfermedad hepática) en el pasado o si consume alcohol con regularidad, ya que se debe tener especial precaución en estos casos.
Durante el tratamiento con este medicamento, el azúcar en sangre (concentración de azúcar en sangre) puede aumentar y, en algunos pacientes con alto riesgo de desarrollar diabetes, puede provocar un aumento excesivo del azúcar en sangre (hiperglucemia), por lo que se requiere tratamiento antidiabético.
Su médico lo controlará de cerca si tiene niveles altos de azúcar en sangre, triglicéridos (grasas) y presión arterial alta.
APLACTIN no es adecuado cuando la hipercolesterolemia se debe a niveles altos de colesterol HDL.
Niños y adolescentes
No se recomienda el uso de APLACTIN en pacientes menores de 18 años.
Interacciones ¿Qué medicamentos o alimentos pueden cambiar el efecto de Aplactin?
Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tener que tomar cualquier otro medicamento.
Informe a su médico si está tomando medicamentos llamados fibratos (por ejemplo, gemfibrozil, fenofibrato) y se utilizan para reducir el contenido de grasa (colesterol) en la sangre; No se recomienda el uso concomitante con APLACTIN porque puede aumentar el riesgo de problemas musculares (ver "Advertencias y precauciones"). Su médico realizará pruebas específicas (control de creatina quinasa, niveles de CK).
Tome este medicamento con precaución y hable con su médico si está tomando los siguientes medicamentos:
- Colestiramina o Colestipol (medicamentos utilizados para reducir los niveles de colesterol en sangre). Cuando APLACTIN se toma simultáneamente con estos medicamentos, puede disminuir su efecto terapéutico. APLACTIN debe tomarse 1 hora antes o 4 horas después de la colestiramina o 1 hora antes del colestipol, para no tener una disminución terapéutica de pravastatina (ver sección 3 "Cómo tomar APLACTIN");
- Ciclosporina. La ingesta simultánea de ciclosporina (un medicamento que se usa para prevenir reacciones de rechazo después del trasplante de órganos) y APLACTINA aumenta la exposición del cuerpo al medicamento en aproximadamente 4 veces o más;
- Eritromicina y claritromicina (antibióticos). Estos medicamentos pueden provocar un aumento de la concentración de pravastatina en sangre.
Si está tomando un medicamento utilizado para tratar y prevenir la formación de coágulos de sangre llamado "antagonista de la vitamina K", informe a su médico antes de tomar APLACTIN, ya que el uso de antagonistas de la vitamina K concomitantemente con APLACTIN puede alterar los resultados del análisis. antagonistas de la vitamina K.
Si necesita tomar ácido fusídico oral para tratar una infección bacteriana, debe dejar de usar este medicamento temporalmente. Su médico le dirá cuándo puede reanudar el tratamiento con APLACTIN. La ingesta de APLACTIN con ácido fusídico rara vez puede provocar debilidad muscular, ardor o dolor (rabdomiólisis). Para obtener más información sobre la rabdomiólisis, ver sección 4 "Posibles efectos adversos".
Advertencias Es importante saber que:
Embarazo, lactancia y fertilidad
El embarazo
No tome APLACTIN durante el embarazo (consulte "No tome APLACTIN").
Si durante el tratamiento con APLACTIN se da cuenta de que está embarazada o tiene intención de quedarse embarazada, informe a su médico de inmediato y detenga el tratamiento debido al riesgo potencial para el feto.
Hora de la comida
No tome APLACTIN durante la lactancia porque una pequeña cantidad de pravastatina se excreta en la leche materna (consulte "No tome APLACTIN").
Conducción y uso de máquinas
La influencia de APLACTIN sobre la capacidad para conducir o utilizar máquinas es nula o insignificante. Sin embargo, puede sentirse mareado después de tomar APLACTIN, si esto le sucede, evite conducir o usar máquinas.
APLACTIN contiene lactosa.
Si su médico le ha dicho que padece "intolerancia a ciertos azúcares, consulte con él antes de tomar este medicamento".
Dosis, método y momento de administración Cómo usar Aplactin: Posología
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico o farmacéutico. En caso de duda, consulte a su médico o farmacéutico.
Antes de iniciar la terapia con APLACTIN, el médico descartará causas secundarias de hipercolesterolemia (por ejemplo, hipotiroidismo, diabetes mellitus, ingesta excesiva de grasas con la dieta).
Antes y durante el tratamiento con APLACTIN, debe seguir una dieta para reducir los niveles de grasa en sangre (dieta estándar para reducir los lípidos). Tome APLACTIN por vía oral una vez al día, preferiblemente por la noche, con o sin alimentos.
La duración del tratamiento varía según prescripción médica.
La dosis recomendada varía en los siguientes casos:
- en el tratamiento de la hipercolesterolemia (niveles elevados de grasa en sangre) es de 10 a 40 mg una vez al día. La respuesta se observa en una semana y el efecto completo se alcanza en cuatro semanas. Su médico controlará sus niveles de grasas en sangre y ajustará la dosis según los resultados de las pruebas. La dosis máxima diaria es de 40 mg.
- en prevención cardiovascular (prevención de trastornos cardíacos) es de 40 mg por día.
- en terapia después de un trasplante de órganos, la dosis inicial recomendada es de 20 mg al día si está siendo tratado con medicamentos específicos (terapia inmunosupresora) Su médico le ajustará la dosis hasta un máximo de 40 mg al día.
Uso en niños y adolescentes.
No se recomienda el uso de APLACTIN.
Uso en ancianos
No es necesario ajustar la dosis en los ancianos a menos que existan factores de riesgo predisponentes (consulte "Advertencias y precauciones")
Uso en pacientes con insuficiencia renal o hepática (problemas de riñón o hígado)
Si tiene insuficiencia renal moderada o grave o insuficiencia hepática significativa, la dosis inicial recomendada es de 10 mg al día. Su médico ajustará la dosis según su nivel de grasa.
Terapia simultánea con otros medicamentos.
Si está tomando una resina, es decir, un fármaco secuestrador de ácidos biliares (como colestiramina y colestipol) al mismo tiempo, tome APLACTIN una "hora antes o al menos cuatro horas después de la resina (consulte" Otros medicamentos y APLACTIN ").
Si está tomando ciclosporina (un medicamento utilizado en el trasplante de órganos), con o sin otros medicamentos inmunosupresores, la dosis inicial recomendada de APLACTIN es de 20 mg al día.
El aumento de la dosis a 40 mg debe hacerse con precaución y bajo supervisión médica (ver "Otros medicamentos y APLACTIN").
Sobredosis Qué hacer si ha tomado demasiado Aplactin
En caso de ingestión accidental / ingesta de una sobredosis de APLACTIN, notifique a su médico inmediatamente o acuda al hospital más cercano.
Si tiene más preguntas sobre el uso de este medicamento, consulte a su médico o farmacéutico.
Efectos secundarios ¿Cuáles son los efectos secundarios de Aplactin?
Como todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Principales efectos secundarios:
- dolor muscular (mialgia), dolor articular (artralgia), calambres musculares, debilidad muscular, niveles elevados de una sustancia producida por los músculos (creatina quinasa, CK);
- aumento de los niveles de enzimas hepáticas (transaminasas séricas).
También se han notificado las siguientes reacciones adversas durante los ensayos clínicos y después de la comercialización de APLACTIN:
Efectos adversos poco frecuentes (pueden afectar hasta 1 de cada 100 personas)
- mareos, dolor de cabeza (dolor de cabeza);
- alteraciones del sueño que incluyen insomnio y pesadillas;
- alteraciones de la visión, que incluyen visión borrosa y visión doble;
- trastornos digestivos y ardientes, dolor abdominal, náuseas, vómitos, estreñimiento, diarrea, hinchazón;
- picazón, sarpullido, urticaria, cambios en el cuero cabelludo y el cabello, incluida la caída del cabello;
- dificultad para orinar (disuria), paso frecuente de pequeñas cantidades de orina (polaquiuria) y necesidad repetida de orinar durante el descanso nocturno (nicturia);
- disfunciones sexuales;
- fatiga.
Efectos adversos muy raros (pueden afectar hasta 1 de cada 10.000 pacientes)
- polineuropatía periférica (enfermedad que afecta a múltiples nervios), especialmente con tratamiento prolongado, y parestesia (disminución de la sensibilidad de una parte del cuerpo);
- reacciones de hipersensibilidad tales como anafilaxia, angioedema, síndrome similar al lupus eritematoso;
- pancreatitis (inflamación del páncreas).
- ictericia (coloración amarillenta de la piel), hepatitis (inflamación del hígado) y necrosis hepática fulminante;
- rabdomiólisis (enfermedad caracterizada por daño a las fibras musculares) que puede estar asociada con insuficiencia renal aguda secundaria a mioglobinuria (presencia de mioglobina en la orina) y miopatía (trastornos musculares) (ver "Advertencias y precauciones").
Efectos secundarios aislados
- trastornos de los tendones, a veces complicados por rotura.
Efectos indeseables de frecuencia desconocida (cuya frecuencia no puede determinarse a partir de los datos disponibles)
- debilidad muscular constante (miopatía necrotizante inmunomediada).
Efectos indeseables relacionados con la clase de estatinas
- pesadillas
- pérdida de memoria;
- depresión;
- enfermedad pulmonar intersticial (enfermedad caracterizada por la alteración del tejido que recubre los alvéolos pulmonares que puede manifestarse con problemas respiratorios como tos persistente y / o dificultad para respirar), en casos excepcionales, especialmente en terapias a largo plazo (ver "Advertencias y precauciones" );
- diabetes mellitus, la frecuencia depende de la presencia o ausencia de factores de riesgo (glucosa en sangre en ayunas ≥ 5,6 mmol / L, IMC> 30 kg / m2, niveles elevados de triglicéridos, antecedentes de hipertensión);
- dermatomiositis (una condición caracterizada por "inflamación de los músculos y la piel).
El cumplimiento de las instrucciones contenidas en el prospecto reduce el riesgo de reacciones adversas.
Notificación de efectos secundarios
Si experimenta cualquier efecto adverso, consulte a su médico o farmacéutico, incluido cualquier posible efecto adverso no mencionado en este prospecto. También puede informar los efectos secundarios directamente a través del sistema nacional de notificación en http://www.agenziafarmaco.gov.it/it/responsabili Al notificar los efectos secundarios, puede ayudar a proporcionar más información sobre la seguridad de este medicamento.
Caducidad y retención
Mantenga este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que se indica en el paquete después de "CAD".
La fecha de caducidad se refiere al último día de ese mes y al producto en envase intacto, correctamente almacenado.
Almacene el medicamento por debajo de 30 ° C.
Conservar en el embalaje original.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que no necesita, ya que esto ayudará a proteger el medio ambiente.
Fecha límite "> Otra información
¿Qué contiene APLACTIN?
APLACTIN 20 mg comprimidos
Una tableta de 20 mg contiene:
el ingrediente activo es la sal sódica de pravastatina 20 mg.
Los demás componentes son lactosa monohidrato, polivinilpirrolidona, celulosa microcristalina, croscarmelosa de sodio, estearato de magnesio, óxido de magnesio, óxido de hierro amarillo (E172).
APLACTIN 40 mg comprimidos
Una tableta de 40 mg contiene:
el ingrediente activo es la sal sódica de pravastatina 40 mg.
Los demás componentes son lactosa monohidrato, polivinilpirrolidona, celulosa microcristalina, croscarmelosa de sodio, estearato de magnesio, óxido de magnesio, óxido de hierro amarillo (E172).
Descripción del aspecto de APLACTIN y contenido del envase
APLACTIN 20 mg comprimidos
Cada paquete contiene 10 comprimidos de 20 mg.
APLACTIN 40 mg comprimidos
Cada paquete contiene 14 comprimidos de 40 mg.
Prospecto fuente: AIFA (Agencia Italiana de Medicamentos). Contenido publicado en enero de 2016. Es posible que la información presente no esté actualizada.
Para tener acceso a la versión más actualizada, es recomendable acceder al sitio web de la AIFA (Agencia Italiana de Medicamentos). Descargo de responsabilidad e información útil.
01.0 NOMBRE DEL MEDICAMENTO -
APLACTIN 20 MG COMPRIMIDOS
02.0 COMPOSICIÓN CUALITATIVA Y CUANTITATIVA -
Cada comprimido contiene 20 mg de sal sódica de pravastatina.
Excipientes con efectos conocidos: lactosa monohidrato.
Para consultar la lista completa de excipientes, ver sección 6.1.
03.0 FORMA FARMACÉUTICA -
Tableta.
04.0 INFORMACIÓN CLÍNICA -
04.1 Indicaciones terapéuticas -
APLACTIN está indicado en adultos en los siguientes casos:
Hipercolesterolemia
Tratamiento de la hipercolesterolemia primaria o dislipidemia mixta, además de la dieta, cuando la respuesta a la dieta u otros tratamientos no farmacológicos (por ejemplo, ejercicio, reducción de peso) ha sido inadecuada.
Prevención primaria
Reducción de la mortalidad y morbilidad cardiovascular en pacientes con hipercolesterolemia de moderada a grave y con alto riesgo de un primer episodio cardiovascular, como complemento de la dieta (ver sección 5.1).
Prevención secundaria
Reducción de la mortalidad y morbilidad cardiovascular en pacientes con antecedentes de infarto de miocardio o angina de pecho inestable y con niveles de colesterol normales o elevados, como complemento de la corrección de otros factores de riesgo (ver sección 5.1).
Postrasplante
Reducción de la hiperlipidemia postrasplante en pacientes sometidos a terapia inmunosupresora tras un trasplante de órganos sólidos (ver secciones 4.2, 4.5 y 5.1).
04.2 Posología y forma de administración -
Antes de iniciar el tratamiento con APLACTIN, deben excluirse las causas secundarias de hipercolesterolemia y los pacientes deben recibir una dieta estándar para reducir los lípidos que se continuará durante el tratamiento.
APLACTIN se administra por vía oral una vez al día, preferiblemente por la noche, con o sin alimentos.
Hipercolesterolemia: el rango de dosificación recomendado es de 10 & -; 40 mg en una sola administración diaria. La respuesta terapéutica es evidente en una semana y el efecto completo de una determinada dosis se obtiene en cuatro semanas, por lo que se deben realizar evaluaciones periódicas del perfil lipídico. y la dosis debe ajustarse en consecuencia.La dosis máxima diaria es de 40 mg.
Prevención cardiovascular: En todos los ensayos clínicos de prevención de la morbilidad y la mortalidad, la única dosis de inicio y mantenimiento estudiada fue de 40 mg al día.
Posología después del trasplante: En pacientes en tratamiento inmunosupresor después de un trasplante de órganos, se recomienda una dosis inicial de 20 mg al día (ver sección 4.5).
Según la respuesta de los parámetros lipídicos, la dosis se puede ajustar hasta 40 mg bajo estrecha supervisión médica (ver sección 4.5).
Niños: La documentación sobre la eficacia y seguridad en pacientes menores de 18 años es limitada, por lo que no se recomienda el uso de APLACTIN en estos pacientes.
Pacientes de edad avanzada: No es necesario ajustar la dosis en estos pacientes a menos que existan factores de riesgo predisponentes (ver sección 4.4).
Insuficiencia renal o hepática: en pacientes con insuficiencia renal moderada o grave o con insuficiencia hepática significativa, se recomienda una dosis inicial de 10 mg al día. La dosis debe ajustarse de acuerdo con la respuesta de los parámetros lipídicos y bajo supervisión médica.
Terapia concomitante: Los efectos hipolipemiantes de APLACTIN sobre el colesterol total y el colesterol LDL aumentan cuando se administra en combinación con una resina secuestradora de ácidos biliares (p. Ej., Colestiramina, colestipol). APLACTIN debe administrarse 1 hora antes o al menos 4 horas después de la resina (ver sección 4.5).
Para los pacientes que reciben ciclosporina, con o sin otros medicamentos inmunosupresores, el tratamiento debe comenzar con pravastatina 20 mg una vez al día y el aumento de la dosis hasta 40 mg debe implementarse con precaución (ver sección 4.5).
04.3 Contraindicaciones -
- Hipersensibilidad al principio activo oa alguno de los excipientes incluidos en la sección 6.1.
- Hepatopatías activas que incluyen elevaciones persistentes e inexplicables de las transaminasas séricas que superan 3 veces el límite superior de la normalidad (ver sección 4.4).
- Embarazo y lactancia (ver sección 4.6).
04.4 Advertencias especiales y precauciones de uso apropiadas -
No se ha evaluado pravastatina en pacientes con hipercolesterolemia familiar homocigótica. El tratamiento no es adecuado cuando la hipercolesterolemia se debe a un colesterol HDL elevado.
Al igual que con otros inhibidores de la HMG-CoA reductasa, no se recomienda la combinación de pravastatina con fibratos (ver sección 4.5).
Trastornos hepáticos: al igual que con otros fármacos hipolipemiantes, se han notificado aumentos moderados de las transaminasas hepáticas. En la mayoría de los casos, los niveles de transaminasas hepáticas volvieron a su valor inicial sin tener que interrumpir el tratamiento. Se debe tener especial cuidado en pacientes que desarrollen elevaciones en los niveles de transaminasas y se debe suspender la terapia si las elevaciones en alanina aminotransferasa (ALT) y aspartato aminotransferasa (AST) exceden 3 veces los límites superiores de la norma y son persistentes.
Se debe tener precaución al administrar pravastatina a pacientes con antecedentes de enfermedad hepática o alcoholismo.
Trastornos musculares: al igual que con otros inhibidores de la HMG-CoA reductasa (estatinas), la pravastatina se ha asociado con la aparición de mialgia, miopatía y, muy raramente, rabdomiólisis. La miopatía debe considerarse en todos los pacientes en tratamiento con estatinas que presenten síntomas musculares de naturaleza desconocida como dolor o tensión, debilidad muscular o calambres musculares. En tales casos, se deben controlar los niveles de creatina quinasa (CK) (ver más abajo). La terapia con estatinas debe suspenderse temporalmente si los niveles de CK son> 5 veces el LSN o en caso de síntomas clínicos graves. Muy raramente (en aproximadamente 1 caso por cada 100.000 pacientes-año), se ha producido rabdomiólisis, con o sin insuficiencia renal secundaria. La rabdomiólisis es una afección aguda, potencialmente mortal, del músculo esquelético que puede desarrollarse en cualquier momento durante el tratamiento y se caracteriza por una destrucción muscular masiva asociada con un aumento sustancial de la CK (generalmente> 30 o 40 veces el límite superior de lo normal) que conduce a mioglobinuria. .
El riesgo de miopatía con el uso de estatinas parece depender de la exposición y, por lo tanto, puede variar con las características individuales del fármaco (debido a las diferencias en la lipofilicidad y la farmacocinética), incluida la dosis y el potencial de interacción farmacológica. Aunque no existe una contraindicación muscular para la prescripción de una estatina, algunos factores predisponentes pueden incrementar el riesgo de toxicidad muscular y por lo tanto justificar una "evaluación cuidadosa de la relación beneficio / riesgo y un seguimiento clínico particular. En estos pacientes, el control de la CK es indicado antes de comenzar el tratamiento con estatinas (ver más abajo).
El riesgo y la gravedad de los trastornos musculares durante la terapia con una estatina aumentan con la coadministración de fármacos que interactúan. En ocasiones, el uso de fibratos solos se asocia con miopatía. Por lo general, debe evitarse el uso combinado de una estatina con fibratos. La coadministración de estatinas y ácido nicotínico debe implementarse con precaución. También se ha descrito un aumento de la incidencia de miopatía en pacientes que reciben otras estatinas en combinación con inhibidores del metabolismo del citocromo P450, lo que puede deberse a interacciones farmacocinéticas que no se han documentado para pravastatina (ver sección 4.5) .Cuando se combina con el tratamiento con estatinas. , los síntomas musculares generalmente se resuelven al suspender el tratamiento.
Niveles de creatina quinasa y su interpretación: No se recomienda la monitorización periódica de la creatincinasa (CK) u otras enzimas musculares en pacientes asintomáticos en tratamiento con estatinas. Sin embargo, se recomienda la monitorización de la CK antes de iniciar la terapia con estatinas en pacientes con factores predisponentes específicos y en pacientes que han desarrollado síntomas musculares durante la terapia con estatinas, como se describe a continuación. Si los niveles basales de CK están significativamente elevados (> 5 veces los límites superiores de lo normal), será necesario volver a medirlos entre 5 y 7 días después para confirmar los resultados.
Una vez medidos, los niveles de CK deben interpretarse en el contexto de otros factores potenciales que pueden causar daño muscular transitorio, como el ejercicio intenso o el trauma muscular.
Antes de iniciar el tratamiento: Se debe tener precaución en pacientes con factores predisponentes como insuficiencia renal, hipotiroidismo, antecedentes de toxicidad muscular con una estatina o con fibratos, antecedentes personales o familiares de trastornos musculares hereditarios o alcoholismo. En estos casos, será necesario medir los niveles de CK antes de comenzar la terapia. También se debe considerar la medición de los niveles de CK antes de iniciar el tratamiento en personas mayores de 70 años, especialmente en presencia de otros factores predisponentes en esta población. Si los niveles basales de CK están significativamente elevados (> 5 veces el límite superior de la normalidad), no se debe iniciar el tratamiento y los niveles deben volver a medirse después de 5 & -; 7 días. Los niveles basales de CK también pueden ser útiles como referencia en caso de un aumento posterior durante el tratamiento con estatinas.
Durante el tratamiento: Se debe advertir a los pacientes que notifiquen de inmediato la aparición de dolor muscular, tensión, debilidad o calambres de naturaleza desconocida. En estos casos, deben medirse los niveles de CK. Si se detecta un nivel de CK notablemente elevado (> 5 veces el límite superior de lo normal), se debe suspender el tratamiento con estatinas. Además, se debe considerar la interrupción del tratamiento si los síntomas musculares son graves y causan molestias durante el día, incluso si el aumento de CK persiste. 5 veces los límites superiores de la norma. Si los síntomas se resuelven y los niveles de CK vuelven a la normalidad, se puede considerar la reintroducción de la terapia con estatinas a una dosis más baja y la vigilancia de cerca. Si se sospecha un trastorno muscular hereditario en estos pacientes, no se recomienda reintroducir la terapia con estatinas.
Ha habido informes muy raros de miopatía necrotizante inmunomediada (IMNM) durante o después del tratamiento con algunas estatinas. La IMNM se caracteriza clínicamente por debilidad muscular proximal persistente y creatina cinasa sérica elevada, que persisten a pesar de la interrupción del tratamiento con estatinas.
Pravastatina no debe coadministrarse con formulaciones de ácido fusídico sistémico o dentro de los 7 días posteriores a la interrupción del tratamiento con ácido fusídico. En pacientes en los que el uso sistémico de ácido fusídico se considera esencial, se debe suspender el tratamiento con estatinas durante la duración del tratamiento con ácido fusídico. Se han notificado casos de rabdomiólisis (incluidas algunas muertes) en pacientes tratados con ácido fusídico y estatinas en combinación (ver sección 4.5). Se debe advertir al paciente que consulte a un médico inmediatamente si experimenta síntomas de debilidad, dolor o sensibilidad muscular.
La terapia con estatinas puede reiniciarse siete días después de que se administre la última dosis de ácido fusídico.
En circunstancias excepcionales, cuando se requiera un tratamiento sistémico prolongado con ácido fusídico, por ejemplo en el caso de infecciones graves, la necesidad de la coadministración de pravastatina y ácido fusídico solo debe considerarse caso por caso y bajo estricto control médico. supervisión.
Enfermedad pulmonar intersticial: se han notificado casos excepcionales de enfermedad pulmonar intersticial con algunas estatinas, especialmente con terapia a largo plazo (ver sección 4.8). Los síntomas pueden incluir disnea, tos no productiva y deterioro de la salud general (fatiga, pérdida de peso y fiebre). Si se sospecha que un paciente ha desarrollado una enfermedad pulmonar intersticial, se debe suspender el tratamiento con estatinas.
Diabetes mellitus: Cierta evidencia sugiere que las estatinas, como efecto de clase, aumentan la glucosa en sangre y en algunos pacientes, con alto riesgo de desarrollar diabetes, pueden inducir un nivel de hiperglucemia tal que la terapia antidiabética sea apropiada. Sin embargo, este riesgo se ve compensado por la reducción del riesgo vascular con el uso de estatinas y, por lo tanto, no debería ser motivo para interrumpir el tratamiento. Pacientes con riesgo (glucosa en ayunas 5,6 - 6,9 mmol / L, IMC> 30 kg / m², niveles elevados de triglicéridos, hipertensión) deben controlarse tanto clínica como bioquímicamente de acuerdo con las directrices nacionales.
Información importante sobre algunos de los componentes:
El medicamento contiene lactosa, por lo que los pacientes con intolerancia hereditaria a galactosa, deficiencia de lactasa de Lapp o problemas de absorción de glucosa o galactosa no deben tomar este medicamento.
04.5 Interacciones con otros medicamentos y otras formas de interacción -
Fibratos: el uso de fibratos solos se asocia ocasionalmente con miopatía.Se ha encontrado un mayor riesgo de eventos adversos musculares, incluida la rabdomiólisis, cuando se administra en combinación con otras estatinas. Estos acontecimientos adversos no pueden excluirse con el uso de pravastatina, por lo que en general debe evitarse el uso combinado de pravastatina y fibratos (p. Ej., Gemfibrozilo, fenofibrato) (ver sección 4.4). Si esta combinación se considera necesaria, se requiere una monitorización clínica cuidadosa y un control de los niveles de CK para los pacientes en este régimen.
Colestiramina / Colestipol: Cuando se administra de forma concomitante, se observó una disminución de aproximadamente un 40 a un 50% en la biodisponibilidad de pravastatina. La administración de pravastatina 1 hora antes o 4 horas después de la colestiramina o 1 hora antes de colestipol no produjo disminuciones clínicamente significativas en la biodisponibilidad o el efecto terapéutico de pravastatina (ver sección 4.2).
Ciclosporina: la coadministración de pravastatina y ciclosporina conduce a un aumento de aproximadamente 4 veces en la exposición sistémica a pravastatina. Sin embargo, en algunos pacientes el aumento de la exposición a pravastatina puede ser mayor. Se recomienda la monitorización clínica y bioquímica de los pacientes. combinación (ver sección 4.2).
Antagonistas de la vitamina K: al igual que con otros inhibidores de la HMG-CoA reductasa, el inicio del tratamiento o el aumento de la dosis de pravastatina en pacientes tratados concomitantemente con inhibidores de la vitamina K (p. Ej., Warfarina u otros anticoagulantes cumarínicos) puede resultar en un aumento del índice internacional normalizado (o INR, internacional). Relación normalizada). Por otro lado, interrumpir o reducir la dosis de pravastatina puede resultar en una reducción del INR. En estas situaciones, es necesario un seguimiento adecuado del INR.
Los parámetros de biodisponibilidad en estado estacionario de pravastatina no se alteraron tras la administración de warfarina.
Fármacos metabolizados por el citocromo P450: el complejo del citocromo P450 no metaboliza metabólicamente de forma significativa la pravastatina. Es por ello que los fármacos que son metabolizados por el sistema del citocromo P450, o que son inhibidores del mismo, pueden añadirse a un régimen estable de pravastatina sin provocar cambios significativos en los niveles plasmáticos de pravastatina, como se ha observado con otras estatinas. Se ha demostrado específicamente la ausencia de interacción farmacocinética significativa con pravastatina para varias sustancias, en particular aquellas que son sustratos / inhibidores de CYP3A4, como diltiazem, verapamilo, itraconazol, ketoconazol, inhibidores de proteasa, zumo de pomelo e inhibidores de CYP2C9 (p. Ej., Fluconazol).
En uno de los dos estudios de interacción con pravastatina y eritromicina se observó un aumento estadísticamente significativo del AUC (70%) y Cmax (121%) de pravastatina. En un estudio similar con claritromicina se observó un aumento estadísticamente significativo en el AUC (110%) y Cmax (127%) .Aunque estos son cambios menores, se debe tener cuidado al combinar pravastatina con eritromicina o claritromicina.
Ácido fusídico: el riesgo de miopatía, incluida la rabdomiólisis, puede aumentar con la administración concomitante de ácido fusídico sistémico y estatinas. El mecanismo de esta interacción (si es farmacodinámico, farmacocinético o ambos) aún se desconoce. Se han notificado casos de rabdomiólisis (incluidas algunas muertes) en pacientes tratados con esta combinación.
Si se requiere un tratamiento sistémico con ácido fusídico, se debe suspender el tratamiento con pravastatina mientras dure el tratamiento con ácido fusídico. Ver también la sección 4.4.
Otros fármacos: No se observaron diferencias estadísticamente significativas en la biodisponibilidad en los estudios de interacción cuando la pravastatina se administra con ácido acetilsalicílico, antiácidos (tomados 1 hora antes de la pravastatina), ácido nicotínico o probucol.
04.6 Embarazo y lactancia -
Embarazo: Pravastatina está contraindicada durante el embarazo y solo debe administrarse a mujeres en edad fértil cuando el embarazo sea muy improbable y cuando se les haya advertido del riesgo potencial. Si se planea o se establece un embarazo, se debe informar al médico de inmediato y se debe suspender el tratamiento con pravastatina debido al riesgo potencial para el feto.
Lactancia: una pequeña cantidad de pravastatina se excreta en la leche materna, por lo que pravastatina está contraindicada durante la lactancia (ver sección 4.3).
04.7 Efectos sobre la capacidad para conducir y utilizar máquinas.
La influencia de pravastatina sobre la capacidad para conducir o utilizar máquinas es nula o insignificante. Sin embargo, al conducir vehículos o utilizar máquinas, debe tenerse en cuenta que pueden producirse mareos durante el tratamiento.
04.8 Efectos indeseables -
La frecuencia de las reacciones adversas se clasifica según la siguiente convención: muy frecuentes (≥1 / 10); común (≥1 / 100,
Ensayos clínicos: APLACTIN se estudió a una dosis de 40 mg en siete ensayos clínicos aleatorizados, doble ciego, controlados con placebo en los que participaron más de 21.000 pacientes tratados con pravastatina (N = 10.764) o placebo (N = 10.719), lo que representa más de 47.000 pacientes. años de exposición a pravastatina. Se siguió a más de 19.000 pacientes durante una mediana de 4,8 y -; 5,9 años.
Se han informado las siguientes reacciones adversas a los medicamentos; ninguno ocurrió con una frecuencia superior al 0,3% en el grupo de pravastatina en comparación con el grupo de placebo.
Trastornos del sistema nervioso:
Poco frecuentes: mareos, dolor de cabeza, alteraciones del sueño, incluidos insomnio y pesadillas.
Trastornos oculares:
Poco frecuentes: alteraciones visuales (incluyendo visión borrosa y diplopía).
Desórdenes gastrointestinales:
Poco frecuentes: dispepsia / ardor, dolor abdominal, náuseas / vómitos, estreñimiento, diarrea, flatulencia.
Trastornos de la piel y del tejido subcutáneo:
Poco frecuentes: prurito, erupción cutánea, urticaria, cambios en el cuero cabelludo / cabello (incluida la alopecia).
Trastornos renales y urinarios.:
Poco frecuentes: alteraciones de la micción (incluyendo disuria, polaquiuria, nicturia)
Enfermedades del aparato reproductor y la mama.:
Poco frecuentes: disfunción sexual.
Desordenes generales y condiciones administrativas del sitio:
Poco frecuentes: fatiga.
Eventos de especial interés clínico
Trastornos musculoesqueléticos y del tejido conjuntivoEn ensayos clínicos se han informado efectos sobre el músculo esquelético, por ejemplo: dolor musculoesquelético que incluye artralgia, calambres musculares, mialgia, debilidad muscular y niveles elevados de CK. El porcentaje de mialgia (1,4% de pravastatina frente a 1,4% de placebo) y debilidad muscular (0,1% de pravastatina frente a 3 veces el LSN y> 10 veces el LSN en CARE, WOSCOPS y LIPIDs son similares en los grupos de placebo (1,6% de pravastatina frente a 1,6%) placebo y pravastatina al 1,0% frente a placebo al 1,0%, respectivamente) (ver sección 4.4).
Trastornos hepatobiliares: se han notificado elevaciones de las transaminasas séricas. En los tres ensayos clínicos a largo plazo controlados con placebo, CARE, WOSCOPS y LIPID, se produjeron alteraciones marcadas en los niveles de ALT y AST (> 3 veces el LSN) con una frecuencia similar (≤ 1,2%) en ambos grupos de tratamiento.
Experiencia poscomercialización
Además de lo anterior, se han notificado las siguientes reacciones adversas desde la comercialización de pravastatina:
Trastornos del sistema nervioso
Muy raras: polineuropatía periférica, especialmente después de un uso prolongado, parestesia.
Trastornos del sistema inmunológico.
Muy raras: reacciones de hipersensibilidad: anafilaxia, angioedema, síndrome similar al lupus eritematoso
Desórdenes gastrointestinales
Muy raras: pancreatitis.
Trastornos hepatobiliares
Muy raras: ictericia, hepatitis, necrosis hepática fulminante.
Trastornos de la piel y del tejido subcutáneo
Dermatomiositis
Trastornos musculoesqueléticos y del tejido conjuntivo
Muy raras: rabdomiólisis que puede estar asociada con insuficiencia renal aguda secundaria a mioglobinuria, miopatía (ver sección 4.4).
Casos aislados de trastornos de los tendones, a veces complicados por rotura.
Frecuencia no conocida: miopatía necrotizante inmunomediada (ver sección 4.4).
También se han informado los siguientes efectos secundarios con el uso de estatinas:
Efectos de clase
• Pesadillas
• Pérdida de memoria
• Depresión
• casos excepcionales de enfermedad pulmonar intersticial, especialmente en el tratamiento a largo plazo (ver sección 4.4)
• Diabetes mellitus: la frecuencia depende de la presencia o ausencia de factores de riesgo (glucemia en ayunas ≥ 5,6 mmol / L, IMC> 30 kg / m², niveles elevados de triglicéridos, antecedentes de hipertensión).
Notificación de sospechas de reacciones adversas
La notificación de sospechas de reacciones adversas que se produzcan después de la autorización del medicamento es importante, ya que permite un seguimiento continuo de la relación beneficio / riesgo del medicamento. Se ruega a los profesionales sanitarios que notifiquen cualquier sospecha de reacciones adversas a través del sistema nacional de notificación. "Dirección http / www.agenziafarmaco.gov.it / it / responsable
04.9 Sobredosis -
Hasta la fecha, la experiencia con la sobredosis de pravastatina es limitada. En caso de sobredosis no existe un tratamiento específico. En este caso, el paciente debe ser tratado sintomáticamente y con las medidas de soporte adecuadas.
05.0 PROPIEDADES FARMACOLÓGICAS -
05.1 "Propiedades farmacodinámicas -
Grupo farmacoterapéutico: agentes reductores de lípidos, agentes reductores de colesterol y triglicéridos, inhibidores de la HMG-CoA reductasa; código ATC: C10AA03
Mecanismo de acción
La pravastatina es un inhibidor competitivo de la 3-hidroxi-3-metil-glutaril coenzima A (HMG-CoA) reductasa, la enzima que cataliza el paso temprano que limita la tasa de biosíntesis del colesterol y produce su efecto hipolipemiante de dos formas Primero , a través de una inhibición competitiva específica y reversible de la HMG-CoA reductasa, la pravastatina produce una modesta reducción en la síntesis de colesterol intracelular. Esto conduce a un aumento en el número de receptores de LDL en la superficie celular y aumenta el catabolismo mediado por receptores y la eliminación del colesterol LDL circulante.
En segundo lugar, la pravastatina inhibe la producción de LDL al inhibir la síntesis hepática de colesterol VLDL, el precursor del colesterol LDL.
Tanto en sujetos sanos como en pacientes con hipercolesterolemia, la sal sódica de pravastatina redujo los siguientes valores de lípidos: colesterol total, colesterol LDL, apolipoproteína B, colesterol VLDL y triglicéridos; mientras que el colesterol HDL y la apolipoproteína A estaban elevados.
Eficacia clínica
Prevención primaria
El West of Scotland Coronary Prevention Study (WOSCOPS) fue un estudio clínico aleatorizado, doble ciego y controlado con placebo de 6.595 pacientes varones de 45 a 64 años con hipercolesterolemia de moderada a grave (colesterol LDL = 155 & -; 232 mg / dl [ 4,0 & -; 6,0 mmol / l]) y sin antecedentes de infarto de miocardio, tratados durante una duración media de 4,8 años con una dosis de 40 mg una vez al día de pravastatina o placebo además de la dieta. En pacientes tratados con pravastatina, los resultados mostraron:
- una reducción del riesgo de muerte por enfermedad coronaria e infarto de miocardio no mortal (la RRR de reducción del riesgo relativo fue del 31%; p = 0,0001 con un riesgo absoluto del 7,9% en el grupo placebo y del 5,5% en los pacientes tratados con pravastatina); los efectos sobre la incidencia de estos eventos cardiovasculares acumulativos ya eran evidentes después de seis meses de tratamiento;
- una reducción en el número total de muertes por un evento cardiovascular (RRR 32%; p = 0,03);
- que teniendo en cuenta los factores de riesgo, también se observa una RRR de muerte por todas las causas del 24% (p = 0,039) entre los pacientes tratados con pravastatina;
- una reducción del riesgo relativo de someter al paciente a procedimientos de revascularización miocárdica (injerto de derivación coronaria o angioplastia coronaria) del 37% (p = 0,009) y de la coronariografía en un 31% (p = 0,007).
El beneficio del tratamiento, de acuerdo con los criterios anteriores, no se conoce en pacientes mayores de 65 años, ya que no pudo ser incluido en el estudio.
Dada la falta de datos sobre pacientes con hipercolesterolemia asociada con niveles de triglicéridos superiores a 5,3 g / l (6 mmol / l) después de una dieta de 8 semanas, no se estableció el beneficio del tratamiento con pravastatina en este tipo de pacientes.
Prevención secundaria
La Intervención a largo plazo con pravastatina en la enfermedad isquémica (LIPID) fue un estudio multicéntrico, aleatorizado, doble ciego y controlado con placebo que comparó los efectos de pravastatina (40 mg una vez al día) con placebo en 9.014 pacientes, entre 31 y 75 años de edad. edad, durante una duración media de 5,6 años, con colesterol normal a alto (colesterol total basal = 155 & -; 271 mg / dl [4,0 & -; 7,0 mmol / l], colesterol total medio = 219 mg / dl [5,66 mmol / l]) y con niveles variables de triglicéridos de hasta 443 mg / dl [5,0 mmol / l] y con antecedentes de infarto de miocardio o angina de pecho inestable en los 3 & -; 36 meses anteriores. El tratamiento con pravastatina redujo significativamente el riesgo relativo de muerte por enfermedad coronaria en un 24% (p = 0,0004, con un riesgo absoluto del 6,4% en el grupo placebo y del 5,3% en pacientes tratados con pravastatina), el riesgo relativo de eventos coronarios ( muerte por enfermedad arterial coronaria o infarto de miocardio no fatal) del 24% (p
- una reducción del riesgo relativo de muerte por todas las causas en un 23% (p cardiovascular en un 25% (p
- una reducción del riesgo relativo de utilizar procedimientos de revascularización miocárdica (injerto de derivación de arterias coronarias o angioplastia coronaria transluminal percutánea) del 20% (p
- una reducción del 19% en el riesgo relativo de accidente cerebrovascular (p = 0,048).
El estudio "Cholesterol and Recurrent Events" (CARE) fue un estudio aleatorizado, doble ciego y controlado con placebo que comparó los efectos de la pravastatina (40 mg una vez al día) sobre la muerte por enfermedad coronaria e infarto de miocardio. media de 4,9 años en 4.159 pacientes, de 21 a 75 años, con colesterol total normal (colesterol total basal medio
- la incidencia de episodios coronarios recurrentes (muerte por enfermedad coronaria o infarto de miocardio no mortal) del 24% (p = 0,003, placebo 13,3%, pravastatina 10,4%);
- el riesgo relativo de recurrir a procedimientos de revascularización (injerto de derivación coronaria o angioplastia coronaria transluminal percutánea) del 27% (p
El riesgo relativo de accidente cerebrovascular también se redujo en un 32% (p = 0,032) y el riesgo de accidente cerebrovascular o ataque isquémico transitorio (AIT) combinado se redujo en un 27% (p = 0,02).
Se desconoce el beneficio del tratamiento según los criterios anteriores en pacientes mayores de 75 años, ya que no pudo incluirse en los estudios CARE y LIPID.
En ausencia de datos sobre pacientes hipercolesterolémicos con niveles de triglicéridos superiores a 3,5 g / l (4 mmol / l) o superiores a 4,45 g / l (5 mmol / l) respectivamente en los estudios CARE y LIPID, después de 4-8 régimen dietético de una semana, no se ha establecido el beneficio del tratamiento con pravastatina en estos pacientes.
En los ensayos clínicos CARE y LIPID, aproximadamente el 80% de los pacientes tomaron ácido acetilsalicílico como parte de su tratamiento.
Trasplante de corazón y riñón
La eficacia de pravastatina en pacientes tratados con inmunosupresores es la siguiente:
• trasplante de corazón, se evaluó en un estudio prospectivo, aleatorizado y controlado (n = 97). Los pacientes fueron tratados simultáneamente con pravastatina (20 & -; 40 mg) o menos y un régimen inmunosupresor estándar de ciclosporina, prednisona y azatioprina. El tratamiento con pravastatina redujo significativamente la incidencia de rechazo cardíaco con compromiso hemodinámico al año, aumentó la supervivencia a un año (p = 0,025) y redujo el riesgo de enfermedad vascular coronaria durante el trasplante, como lo demostró la angiografía y la autopsia (p = 0,049 ).
• Trasplante de riñón, se evaluó en un estudio prospectivo, no controlado, no aleatorizado (n = 48) de 4 meses de duración. Los pacientes fueron tratados simultáneamente con pravastatina (20 mg) o menos y un régimen inmunosupresor estándar de ciclosporina y prednisona.
En pacientes con trasplante renal, pravastatina redujo significativamente tanto la incidencia de episodios de rechazo múltiple, la incidencia de episodios de rechazo agudo confirmados por biopsia como el uso de inyecciones intermitentes tanto de prednisolona como de Muromonab-CD3.
05.2 "Propiedades farmacocinéticas -
Absorción
La pravastatina se administra por vía oral en su forma activa. Se absorbe rápidamente y los niveles séricos máximos se alcanzan 1 & -; 1,5 horas después de la ingestión. En promedio, se absorbe el 34% de la dosis oral, con una biodisponibilidad absoluta del 17%.
La presencia de alimentos en el tracto gastrointestinal conduce a una reducción de la biodisponibilidad, pero el efecto reductor del colesterol de la pravastatina es el mismo si se toma con o sin alimentos.
Después de la absorción, el 66% de la pravastatina se somete a una primera extracción de la circulación en el hígado, que es el sitio principal de su acción y el sitio principal de síntesis de colesterol y eliminación del colesterol LDL. Estudios in vitro han demostrado que la pravastatina se transporta a los hepatocitos y, en un grado sustancialmente menor, a otras células.
A la luz de este primer paso sustancial a través del hígado, las concentraciones plasmáticas de pravastatina tienen un valor limitado para predecir un efecto hipolipemiante.
Las concentraciones plasmáticas son proporcionales a las dosis administradas.
Distribución
Aproximadamente el 50% de la pravastatina circulante se une a las proteínas plasmáticas.
El volumen de distribución es de aproximadamente 0,5 l / kg.
Una pequeña cantidad de pravastatina pasa a la leche materna.
Metabolismo y eliminación
La pravastatina no es metabolizada significativamente por el citocromo P450 ni parece ser un sustrato o inhibidor de la glicoproteína P, sino un sustrato de otras proteínas de transporte.
Después de la administración oral, el 20% de la dosis inicial se elimina en la orina y el 70% en las heces. La vida media de eliminación plasmática de pravastatina oral es de una hora y media a dos horas.
Después de la administración intravenosa, el 47% de la dosis se elimina por excreción renal y el 53% por excreción biliar y biotransformación. El principal producto de degradación de la pravastatina es el metabolito 3-α-hidroxi isomérico. Este metabolito posee una décima a una cuadragésima parte de la actividad inhibidora de la HMG-CoA reductasa del compuesto original.
El aclaramiento sistémico de pravastatina es de 0,81 l / h / kg y el aclaramiento renal es de 0,38 l / h / kg, lo que indica secreción tubular.
Poblaciones en riesgo
Insuficiencia hepática: La exposición sistémica a pravastatina y sus metabolitos en pacientes con cirrosis alcohólica aumenta aproximadamente en un 50% en comparación con pacientes con función hepática normal.
Insuficiencia renal: no se encontraron cambios significativos en pacientes con insuficiencia renal leve. Sin embargo, la insuficiencia renal grave y moderada puede conducir a un aumento del doble de la exposición sistémica a pravastatina y sus metabolitos.
05.3 Datos preclínicos sobre seguridad -
Según estudios convencionales de farmacología de seguridad, toxicidad a dosis repetidas y toxicidad reproductiva, no existen otros riesgos para el paciente que los esperados debido al mecanismo de acción farmacológico.
Los estudios de dosis repetidas indican que la pravastatina puede inducir diversos grados de hepatotoxicidad y miopatía; en general, los efectos sustanciales en estos tejidos solo fueron evidentes a dosis 50 veces o más de la dosis máxima humana en mg / kg.
En los estudios in vitro y en vivo de toxicología genética, no se encontró evidencia de potencial mutagénico.
En un estudio de carcinogenicidad de 2 años en ratones que utilizó pravastatina en dosis de 250 y 500 mg / kg / día (≥ 310 veces la dosis máxima humana en mg / kg), aumentos estadísticamente significativos en la incidencia de carcinomas hepatocelulares en machos y hembras y , solo en mujeres, de adenomas pulmonares. En un estudio de carcinogenicidad de 2 años en ratas, a una dosis de 100 mg / kg / día (125 veces la dosis máxima en humanos en mg / kg), se demostró un aumento estadísticamente significativo en la incidencia de carcinomas hepatocelulares, solo en machos.
06.0 INFORMACIÓN FARMACÉUTICA -
06.1 Excipientes -
Lactosa monohidrato, polivinilpirrolidona, celulosa microcristalina, croscarmelosa de sodio, estearato de magnesio, óxido de magnesio, óxido de hierro amarillo (E172).
06.2 Incompatibilidad "-
Irrelevante.
06.3 Período de validez "-
2 años.
06.4 Precauciones especiales de conservación
Almacenar a una temperatura no superior a 30 ° C.
Almacenar en el envase original
06.5 Naturaleza del envase primario y contenido del envase.
Blister que contiene 10 comprimidos de 20 mg.
06.6 Instrucciones de uso y manipulación -
Sin instrucciones especiales.
07.0 TITULAR DE LA "AUTORIZACIÓN DE COMERCIALIZACIÓN" -
FIRMA. S.p.A., Via di Scandicci 37 - Florencia
08.0 NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN -
A.I.C. N ° 027786021
09.0 FECHA DE LA PRIMERA AUTORIZACIÓN O RENOVACIÓN DE LA AUTORIZACIÓN -
Fecha de la primera autorización: 1 de marzo de 1993
Fecha de la renovación más reciente: 1 de marzo de 2008
10.0 FECHA DE REVISIÓN DEL TEXTO -
Mayo de 2016