Ingredientes activos: Epoetin alfa
Binocrit 1.000 UI / 0,5 ml solución inyectable en jeringa precargada.
Binocrit 2000 UI / 1 ml solución inyectable en jeringa precargada.
Binocrit 3000 UI / 0,3 ml solución inyectable en jeringa precargada.
Binocrit 4000 UI / 0,4 ml solución inyectable en jeringa precargada.
Binocrit 5000 UI / 0,5 ml solución inyectable en jeringa precargada.
Binocrit 6000 UI / 0,6 ml solución inyectable en jeringa precargada.
Binocrit 7.000 UI / 0,7 ml solución inyectable en jeringa precargada.
Binocrit 8.000 UI / 0,8 ml solución inyectable en jeringa precargada.
Binocrit 9.000 UI / 0,9 ml solución inyectable en jeringa precargada.
Binocrit 10.000 UI / 1 ml solución inyectable en jeringa precargada.
Binocrit 20.000 UI / 0,5 ml solución inyectable en jeringa precargada.
Binocrit 30.000 UI / 0,75 ml solución inyectable en jeringa precargada.
Binocrit 40.000 UI / 1 ml solución inyectable en jeringa precargada.
Indicaciones ¿Por qué se usa Binocrit? ¿Para qué sirve?
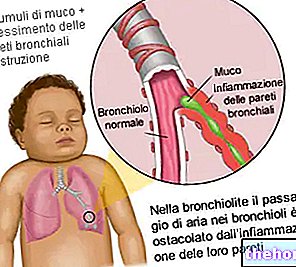
Binocrit contiene el principio activo epoetina alfa, una proteína que estimula la médula ósea para producir más glóbulos rojos que transportan hemoglobina (una sustancia que transporta oxígeno).
La epoetina alfa es una copia de la proteína humana eritropoyetina y funciona de la misma manera.
Binocrit se utiliza para tratar la anemia sintomática causada por una enfermedad renal:
- en niños sometidos a hemodiálisis
- en adultos sometidos a hemodiálisis o diálisis peritoneal,
- en adultos gravemente anémicos que aún no reciben diálisis.
Si tiene una enfermedad renal, es posible que tenga una escasez de glóbulos rojos si el riñón no produce suficiente eritropoyetina (que es necesaria para la producción de glóbulos rojos). Binocrit se prescribe para estimular la médula ósea para que produzca más glóbulos rojos.
Binocrit se utiliza para tratar la anemia cuando está recibiendo quimioterapia para tumores sólidos, linfoma maligno o mieloma múltiple (cáncer de médula ósea) y si su médico decide que puede necesitar una transfusión de sangre. Binocrit puede reducir la necesidad de transfusiones de sangre.
Binocrit se usa en personas con anemia moderada que donan parte de su sangre antes de la cirugía para que la sangre recolectada se les pueda administrar durante o después de la cirugía. Debido a que Binocrit estimula la producción de glóbulos rojos, los médicos pueden extraer más sangre de estas personas.
Binocrit se usa en adultos con anemia moderada que están a punto de someterse a una cirugía ortopédica mayor (como una cirugía de reemplazo de cadera o rodilla) para reducir la posible necesidad de transfusiones de sangre.
Contraindicaciones cuando no se debe utilizar Binocrit
No use Binocrit
- si es alérgico a la epoetina alfa oa cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
- si le han diagnosticado 'aplasia pura de glóbulos rojos (la médula ósea no puede producir suficientes glóbulos rojos) después del tratamiento con cualquier medicamento que estimule la producción de glóbulos rojos (incluido Binocrit) Ver sección 4.
- si tiene la presión arterial alta que no está suficientemente controlada con medicamentos.
- para estimular la producción de glóbulos rojos (para que los médicos puedan extraerle más sangre) si no puede recibir transfusiones con su propia sangre durante o después de la cirugía.
- si está a punto de someterse a una cirugía ortopédica electiva mayor (como una cirugía de cadera o rodilla) y:
- tiene una enfermedad cardíaca grave
- tiene una enfermedad grave de las venas o arterias
- ha tenido recientemente un ataque cardíaco o un derrame cerebral
- no puede tomar medicamentos para diluir la sangre. Es posible que Binocrit no sea adecuado para usted. Discuta esto con su médico.
Algunas personas necesitan medicamentos para reducir el riesgo de coágulos de sangre mientras reciben tratamiento con Binocrit. Si no puede tomar medicamentos que eviten los coágulos de sangre, no debe tomar Binocrit.
Precauciones de uso Lo que necesita saber antes de tomar Binocrit
Hable con su médico, farmacéutico o enfermero antes de usar Binocrit.
Binocrit y otros productos que estimulan la producción de glóbulos rojos pueden aumentar el riesgo de coágulos sanguíneos en todos los pacientes. Este riesgo puede ser mayor si tiene otros factores de riesgo de desarrollar coágulos de sangre (por ejemplo, si ha tenido un coágulo de sangre en el pasado o si tiene sobrepeso, diabetes, enfermedad cardíaca o necesita recostarse por un tiempo. a largo plazo debido a cirugía o enfermedad). Informe a su médico sobre cualquier situación de este tipo. Su médico le ayudará a decidir si Binocrit es adecuado para usted.
Es importante que informe a su médico si se encuentra en alguna de las situaciones siguientes.
Es posible que pueda usar Binocrit de todos modos, pero primero debe hablar con su médico.
Si sabe que tiene dolor o ha sufrido en el pasado de:
- Alta presión sanguínea;
- convulsiones o convulsiones;
- enfermedad del higado;
- anemia por otras causas;
- porfiria (un trastorno sanguíneo raro).
Si es un paciente con cáncer, tenga en cuenta que los medicamentos que estimulan la producción de glóbulos rojos (como Binocrit) pueden actuar como factores de crecimiento y, por lo tanto, teóricamente podrían afectar la progresión del tumor.
Dependiendo de su situación personal, puede ser preferible una transfusión de sangre. Discuta esto con su médico.
Si tiene hepatitis C y recibe interferón y ribavirina, debe comentarlo con su médico porque la combinación de epoetina alfa con interferón y ribavirina ha provocado, en raras ocasiones, una reducción del efecto del tratamiento y una afección denominada aplasia pura de glóbulos rojos ( PRCA), una forma grave de anemia. Binocrit no está aprobado para el tratamiento de la anemia asociada con la hepatitis C.
Si es un paciente con insuficiencia renal crónica, y en particular si no responde adecuadamente a Binocrit, su médico comprobará la dosis de Binocrit que recibe porque aumentar repetidamente la dosis de Binocrit si no responde al tratamiento puede aumentar el riesgo. de problemas del corazón o de los vasos sanguíneos y el riesgo de infarto de miocardio, accidente cerebrovascular y muerte.
Tenga especial cuidado con otros productos que estimulan la producción de glóbulos rojos: Binocrit pertenece a un grupo de productos que, como la proteína humana eritropoyetina, estimulan la producción de glóbulos rojos. Su profesional de la salud siempre anotará el producto específico que está usando.Si le administran algún medicamento de este grupo, que no sea Binocrit, durante su tratamiento, hable con su médico o farmacéutico antes de usarlo.
Interacciones ¿Qué medicamentos o alimentos pueden cambiar el efecto de Binocrit?
Binocrit no suele reaccionar con otros medicamentos, pero informe a su médico si está utilizando, ha utilizado recientemente o podría utilizar cualquier otro medicamento, incluso los adquiridos sin receta.
Si toma un medicamento llamado ciclosporina (utilizado, por ejemplo, después de un trasplante de riñón), es posible que su médico le haga análisis de sangre para medir sus niveles de ciclosporina mientras esté tomando Binocrit.
Los suplementos de hierro y otros estimulantes de la sangre pueden aumentar la eficacia de Binocrit. Su médico decidirá si es adecuado que los tome.
Si acude a un hospital, clínica o médico de familia, dígales que está siendo tratado con Binocrit. Esto podría afectar a otros tratamientos o resultados de pruebas.
Advertencias Es importante saber que:
Embarazo y lactancia
Es importante que informe a su médico si se encuentra en alguna de las situaciones siguientes.
Es posible que pueda usar Binocrit de todos modos, pero primero debe discutir esto con su médico:
- si está embarazada o si sospecha que puede estarlo.
- si está amamantando.
Binocrit contiene sodio
Binocrit contiene menos de 1 mmol (23 mg) de sodio por dosis, es decir, está esencialmente "exento de sodio".
Dosis, método y momento de administración Cómo utilizar Binocrit: Posología
Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte a su médico.
El médico realizó un análisis de sangre y decidió que necesita Binocrit.
Binocrit se puede administrar mediante inyección:
- en una vena o tubo que va a una vena (por vía intravenosa)
- o debajo de la piel (por vía subcutánea).
Su médico decidirá cómo se inyectará Binocrit. Por lo general, las inyecciones las administra un médico, una enfermera u otro profesional de la salud. Algunas personas, según el motivo por el que necesiten tratamiento con Binocrit, pueden aprender más adelante cómo inyectarse debajo de la piel: consulte las Instrucciones para autoinyectarse Binocrit al final de este prospecto.
Binocrit no debe utilizarse:
- después de la fecha de caducidad que se muestra en la etiqueta y el embalaje exterior
- si sabe o cree que se ha congelado accidentalmente o
- si ha ocurrido una falla en el refrigerador.
La dosis de Binocrit que le administrarán depende de su peso corporal en kilogramos. La causa de la anemia también es importante para que el médico elija la dosis correcta.
Su médico controlará su presión arterial con regularidad durante el tratamiento con Binocrit.
Personas con enfermedad renal
- Su médico mantendrá su nivel de hemoglobina entre 10 y 12 g / dL, ya que un nivel alto de hemoglobina podría aumentar el riesgo de coágulos sanguíneos y muerte.
- La dosis inicial habitual de Binocrit en adultos y niños es de 50 Unidades Internacionales (UI) por kilogramo (/ kg) de peso corporal, administradas tres veces por semana. En pacientes sometidos a diálisis peritoneal, Binocrit puede administrarse dos veces por semana.
- En adultos y niños, Binocrit se administra en forma de inyección en una vena (por vía intravenosa) o en un tubo que se introduce en una vena. Cuando este acceso (a través de una vena o tubo) no está disponible, el médico puede decidir inyectar Binocrit debajo de la piel (por vía subcutánea). Esto afecta a los pacientes en diálisis y a los pacientes que aún no reciben diálisis.
- Su médico ordenará análisis de sangre con regularidad para ver cómo responde la anemia a la terapia y puede ajustar la dosis, generalmente cada cuatro semanas a más tardar.
- Una vez que se haya corregido la anemia, su médico seguirá analizándole la sangre con regularidad. La dosis y la frecuencia de administración de Binocrit pueden ajustarse aún más para mantener su respuesta al tratamiento. Su médico utilizará la dosis efectiva más baja para controlar sus síntomas. Anemia .
- Si no responde adecuadamente a Binocrit, su médico comprobará la dosis que está recibiendo y le informará si es necesario cambiar sus dosis de Binocrit.
- Si está utilizando un intervalo más largo (más de una vez a la semana) entre las dosis de Binocrit, es posible que no pueda mantener niveles adecuados de hemoglobina y es posible que deba aumentar la dosis o la frecuencia de administración de Binocrit.
- También puede recibir suplementos de hierro antes y durante el tratamiento con Binocrit para aumentar la eficacia del tratamiento.
- Si está en diálisis cuando comienza el tratamiento con Binocrit, es posible que deba cambiar su horario de diálisis. El médico decidirá sobre esto.
Adultos en quimioterapia
- Su médico puede comenzar la terapia con Binocrit si su hemoglobina es de 10 g / dL o menos.
- Su médico mantendrá su nivel de hemoglobina entre 10 y 12 g / dL, ya que un nivel alto de hemoglobina puede aumentar el riesgo de coágulos sanguíneos y muerte.
- La dosis inicial habitual es de 150 UI por kilogramo de peso corporal tres veces por semana o 450 UI por kilogramo de peso corporal una vez a la semana.
- Binocrit se administra mediante inyección debajo de la piel.
- Su médico ordenará análisis de sangre y puede ajustar la dosis dependiendo de cómo responda su anemia al tratamiento.
- También puede recibir suplementos de hierro antes y durante el tratamiento con Binocrit para aumentar la eficacia del tratamiento.
- El tratamiento con Binocrit generalmente continuará durante un mes después del final de la quimioterapia.
Adultos que donan su propia sangre
- La dosis habitual es de 600 UI por kilogramo de peso corporal, dos veces por semana.
- Binocrit se administra mediante inyección en una vena, inmediatamente después de la donación de sangre, durante 3 semanas antes de la cirugía.
- También puede recibir suplementos de hierro antes y durante el tratamiento con Binocrit para aumentar la eficacia del tratamiento. Adultos programados para cirugía ortopédica mayor
- La dosis recomendada es de 600 UI por kilogramo de peso corporal una vez a la semana.
- Binocrit se administra mediante inyección debajo de la piel todas las semanas durante tres semanas antes de la cirugía y el día de la cirugía.
- Si es necesario acortar el tiempo antes de la cirugía, se administra una dosis diaria de 300 UI / kg durante un máximo de diez días antes de la cirugía, el día de la cirugía y los cuatro días siguientes.
- Si los análisis de sangre muestran valores de hemoglobina demasiado altos antes de la cirugía, se interrumpirá el tratamiento.
- Puede recibir suplementos de hierro antes y durante el tratamiento con Binocrit para aumentar la eficacia del tratamiento.
Instrucciones para inyectar Binocrit debajo de la piel.
Al comienzo del tratamiento, Binocrit generalmente es inyectado por personal médico o paramédico. Posteriormente, su médico puede sugerirle a usted, o un cuidador, que aprenda a inyectarse Binocrit debajo de la piel (por vía subcutánea) por sí solo.
- No intente inyectarse este medicamento usted mismo a menos que su médico o enfermero le hayan enseñado cómo hacerlo.
- Utilice siempre Binocrit exactamente como lo indique su médico o enfermero.
- Asegúrese de inyectar solo la cantidad de líquido indicada por su médico o enfermero.
- Utilice Binocrit solo si se ha almacenado correctamente; consulte la sección 5, Conservación de Binocrit.
- Antes de usar, deje reposar la jeringa de Binocrit hasta que alcance la temperatura ambiente (esto suele tardar entre 15 y 30 minutos) .Use la jeringa dentro de los 3 días posteriores a su extracción del refrigerador.
Tome solo una dosis de Binocrit de cada jeringa.
Si Binocrit se inyecta debajo de la piel (por vía subcutánea), el volumen inyectado generalmente no excede un mililitro (1 ml) por inyección única.
Binocrit se administra solo y no se mezcla con otros líquidos inyectables.
No agite las jeringas de Binocrit. La agitación vigorosa prolongada puede dañar el producto. Si el producto ha sido agitado vigorosamente, no lo utilice.
Las instrucciones de autoinyección de Binocrit se encuentran al final de este prospecto.
Si olvidó usar Binocrit
Administre la siguiente inyección tan pronto como se acuerde. Si falta menos de un día para la próxima inyección, omita la inyección omitida y continúe con su horario normal. No se inyecte una dosis doble.
Si tiene más preguntas sobre el uso de este medicamento, consulte a su médico, enfermero o farmacéutico.
Sobredosis Qué hacer si ha tomado demasiado Binocrit
Informe a su médico o enfermero inmediatamente si cree que se ha inyectado demasiado Binocrit. Es poco probable que se produzcan efectos secundarios si toma una sobredosis de Binocrit.
Efectos secundarios ¿Cuáles son los efectos secundarios de Binocrit?
Como todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Informe a su médico o enfermera de inmediato si nota alguno de los efectos secundarios enumerados.
Efectos secundarios muy frecuentes
Pueden afectar a más de 1 de cada 10 personas que usan Binocrit.
- Diarrea
- Náusea
- Él vomitó
- Fiebre
- Se han notificado casos de congestión del tracto respiratorio, como congestión nasal y dolor de garganta, en pacientes con enfermedad renal que aún no están en diálisis.
Efectos secundarios comunes
Pueden afectar hasta 1 de cada 10 personas que utilizan Binocrit.
- Aumento de la presión arterial. Los siguientes signos pueden indicar un aumento repentino de la presión arterial: dolor de cabeza, especialmente si es de aparición repentina y un tipo punzante similar a la migraña, confusión o convulsiones. Estos signos requieren tratamiento urgente. El aumento de la presión arterial en sangre puede requerir tratamiento con otros medicamentos (o un ajuste en la dosis de los medicamentos que ya está tomando para la presión arterial alta).
- Coágulos de sangre (incluida la trombosis venosa profunda y la embolia) que pueden requerir una intervención urgente. Los síntomas pueden incluir dolor en el pecho, dificultad para respirar e hinchazón con dolor y enrojecimiento, generalmente en las piernas.
- Tos.
- Erupción cutánea, que puede deberse a una reacción alérgica.
- Dolor en los huesos o músculos.
- Síntomas similares a los de la gripe como dolor de cabeza, dolores y molestias en las articulaciones, sensación de debilidad, escalofríos, cansancio y mareos. Estos síntomas pueden ser más comunes al inicio del tratamiento. Si experimenta estos síntomas mientras se inyecta en la vena, una inyección más lenta puede ayudar a evitarlos en el futuro.
- Enrojecimiento, ardor y dolor en el lugar de la inyección.
- Hinchazón de tobillos, pies o dedos.
Efectos secundarios poco frecuentes
Pueden afectar hasta 1 de cada 100 personas que utilizan Binocrit.
- Niveles altos de potasio en sangre, que pueden provocar ritmos cardíacos anormales (este es un efecto adverso muy común en pacientes en diálisis).
- Convulsiones
- Congestión de la nariz o las vías respiratorias.
Efectos secundarios muy raros
Pueden afectar hasta 1 de cada 10.000 personas que utilizan Binocrit.
- Síntomas de la aplasia pura de glóbulos rojos (PRCA) La aplasia pura de glóbulos rojos (PRCA) significa que la médula ósea no produce suficientes glóbulos rojos. La PRCA causa "anemia repentina y grave. Los síntomas son:
- cansancio inusual,
- sintiéndose mareado,
- dificultad para respirar
En muy raras ocasiones, se ha notificado PRCA, especialmente en pacientes con enfermedad renal, después de meses o años de tratamiento con epoetina alfa y otros medicamentos que estimulan la producción de glóbulos rojos.
- Sobre todo al inicio del tratamiento, puede producirse un aumento del número de algunas pequeñas células sanguíneas (llamadas plaquetas), que normalmente están implicadas en la formación de coágulos, por lo que su médico realizará las comprobaciones pertinentes.
Si está en hemodiálisis:
- Se pueden formar coágulos (trombosis) en la fístula de diálisis. Es más probable que esto suceda si tiene la presión arterial baja o si hay complicaciones con la fístula.
- También se pueden formar coágulos en el sistema de hemodiálisis. El médico puede decidir aumentar la dosis de heparina durante la diálisis.
Informe a su médico o enfermero inmediatamente si nota alguno de estos efectos o si nota cualquier otro efecto mientras toma Binocrit.
Si alguno de los efectos adversos se agrava o si nota cualquier efecto adverso no mencionado en este prospecto, comuníqueselo a su médico, enfermero o farmacéutico.
Notificación de efectos secundarios
Si experimenta cualquier efecto adverso, consulte a su médico, farmacéutico o enfermero. Esto incluye cualquier posible efecto adverso que no se mencione en este prospecto. También puede notificar los efectos secundarios directamente a través del sistema nacional de notificación incluido en el Apéndice V.Efectos secundarios que puede ayudar proporcionar más información sobre la seguridad de este medicamento.
Caducidad y retención
- Mantenga este medicamento fuera de la vista y del alcance de los niños.
- No utilice este medicamento después de la fecha de caducidad que aparece en la etiqueta después de "CAD" y en la caja después de "CAD".
- Conservar y transportar refrigerado (entre 2 ° C y 8 ° C).
- Puede sacar Binocrit de la nevera y conservarlo a temperatura ambiente (hasta 25 ° C) durante un máximo de 3 días. Una vez que la jeringa se ha sacado de la nevera y ha alcanzado la temperatura ambiente (hasta 25 ° C), debe utilizarse en los 3 días siguientes o desecharse.
- No congelar ni agitar.
- Conservar en el paquete original para proteger el medicamento de la luz.
No use este medicamento si nota
- que se ha congelado accidentalmente o
- que ha ocurrido una falla en el refrigerador
- que el líquido tiene color o si ve partículas flotando en él
- que el sello está roto.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que no necesita, ya que esto ayudará a proteger el medio ambiente.
Fecha límite "> Otra información
Qué contiene Binocrit
- El ingrediente activo es epoetina alfa (consulte la cantidad en la tabla a continuación).
- Los demás componentes son dihidrogenofosfato de sodio dihidrato, fosfato de disodio dihidrato, cloruro de sodio, glicina, polisorbato 80, ácido clorhídrico (para ajustar el pH), hidróxido de sodio (para ajustar el pH), agua para preparaciones inyectables.
Aspecto de Binocrit y contenido del envase
Binocrit se presenta como una solución inyectable transparente e incolora en una jeringa precargada. Las jeringas están selladas en ampollas.
* Envases de 1, 4 o 6 jeringas precargadas con o sin protector de seguridad para la aguja.
Es posible que no se comercialicen todos los tamaños de envases.
Prospecto fuente: AIFA (Agencia Italiana de Medicamentos). Contenido publicado en enero de 2016. Es posible que la información presente no esté actualizada.
Para tener acceso a la versión más actualizada, es recomendable acceder al sitio web de la AIFA (Agencia Italiana de Medicamentos). Descargo de responsabilidad e información útil.
01.0 NOMBRE DEL MEDICAMENTO -
SOLUCIÓN BINOCRIT PARA INYECCIÓN EN JERINGA PRECARGADA
02.0 COMPOSICIÓN CUALITATIVA Y CUANTITATIVA -
Binocrit 1.000 UI / 0,5 ml solución inyectable en jeringa precargada.
Cada ml de solución contiene 2000 UI de epoetina alfa *, lo que corresponde a 16,8 mcg por ml.
Una jeringa precargada de 0,5 ml contiene 1.000 unidades internacionales (UI), que corresponden a 8,4 mcg de epoetina alfa. *
Binocrit 2000 UI / 1 ml solución inyectable en jeringa precargada.
Cada ml de solución contiene 2000 UI de epoetina alfa *, lo que corresponde a 16,8 mcg por ml.
Una jeringa precargada de 1 ml contiene 2000 unidades internacionales (UI), que corresponden a 16,8 mcg de epoetina alfa. *
Binocrit 3000 UI / 0,3 ml solución inyectable en jeringa precargada.
Cada ml de solución contiene 10.000 UI de epoetina alfa *, lo que corresponde a 84,0 mcg por ml.
Una jeringa precargada de 0,3 ml contiene 3000 unidades internacionales (UI), que corresponden a 25,2 mcg de epoetina alfa. *
Binocrit 4000 UI / 0,4 ml solución inyectable en jeringa precargada.
Cada ml de solución contiene 10.000 UI de epoetina alfa *, lo que corresponde a 84,0 mcg por ml.
Una jeringa precargada de 0,4 ml contiene 4000 unidades internacionales (UI), que corresponden a 33,6 mcg de epoetina alfa. *
Binocrit 5000 UI / 0,5 ml solución inyectable en jeringa precargada.
Cada ml de solución contiene 10.000 UI de epoetina alfa *, lo que corresponde a 84,0 mcg por ml.
Una jeringa precargada de 0,5 ml contiene 5.000 unidades internacionales (UI), que corresponden a 42,0 mcg de epoetina alfa. *
Binocrit 6000 UI / 0,6 ml solución inyectable en jeringa precargada.
Cada ml de solución contiene 10.000 UI de epoetina alfa *, lo que corresponde a 84,0 mcg por ml.
Una jeringa precargada de 0,6 ml contiene 6000 unidades internacionales (UI), que corresponden a 50,4 mcg de epoetina alfa. *
Binocrit 7.000 UI / 0,7 ml solución inyectable en jeringa precargada.
Cada ml de solución contiene 10.000 UI de epoetina alfa *, lo que corresponde a 84,0 mcg por ml.
Una jeringa precargada de 0,7 ml contiene 7.000 unidades internacionales (UI), que corresponden a 58,8 mcg de epoetina alfa. *
Binocrit 8.000 UI / 0,8 ml solución inyectable en jeringa precargada.
Cada ml de solución contiene 10.000 UI de epoetina alfa *, lo que corresponde a 84,0 mcg por ml.
Una jeringa precargada de 0,8 ml contiene 8.000 unidades internacionales (UI), que corresponden a 67,2 mcg de epoetina alfa. *
Binocrit 9.000 UI / 0,9 ml solución inyectable en jeringa precargada.
Cada ml de solución contiene 10.000 UI de epoetina alfa *, lo que corresponde a 84,0 mcg por ml.
Una jeringa precargada de 0,9 ml contiene 9.000 unidades internacionales (UI), que corresponden a 75,6 mcg de epoetina alfa. *
Binocrit 10.000 UI / 1 ml solución inyectable en jeringa precargada.
Cada ml de solución contiene 10.000 UI de epoetina alfa *, lo que corresponde a 84,0 mcg por ml.
Una jeringa precargada de 1 ml contiene 10.000 unidades internacionales (UI), que corresponden a 84,0 mcg de epoetina alfa. *
Binocrit 20.000 UI / 0,5 ml solución inyectable en jeringa precargada.
Cada ml de solución contiene 40.000 UI de epoetina alfa *, lo que corresponde a 336,0 mcg por ml.
Una jeringa precargada de 0,5 ml contiene 20.000 unidades internacionales (UI), que corresponden a 168,0 mcg de epoetina alfa. *
Binocrit 30.000 UI / 0,75 ml solución inyectable en jeringa precargada.
Cada ml de solución contiene 40.000 UI de epoetina alfa *, lo que corresponde a 336,0 mcg por ml.
Una jeringa precargada de 0,75 ml contiene 30.000 unidades internacionales (UI), que corresponden a 252,0 mcg de epoetina alfa. *
Binocrit 40.000 UI / 1 ml solución inyectable en jeringa precargada.
Cada ml de solución contiene 40.000 UI de epoetina alfa *, lo que corresponde a 336,0 mcg por ml.
Una jeringa precargada de 1 ml contiene 40.000 unidades internacionales (UI), correspondientes a 336,0 mcg de epoetina alfa. *
* Producido en células de ovario de hámster chino (CHO) mediante tecnología de ADN recombinante
Para consultar la lista completa de excipientes, ver sección 6.1.
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por dosis, es decir, esencialmente "exento de sodio".
03.0 FORMA FARMACÉUTICA -
Solución inyectable en jeringa precargada (inyección)
Solución transparente e incolora
04.0 INFORMACIÓN CLÍNICA -
04.1 Indicaciones terapéuticas -
Binocrit está indicado para el tratamiento de la anemia sintomática asociada a insuficiencia renal crónica (IRC):
• en adultos y niños en hemodiálisis de 1 a 18 años y pacientes adultos sometidos a diálisis peritoneal (ver sección 4.4).
• en pacientes adultos con insuficiencia renal que aún no se hayan sometido a diálisis para el tratamiento de la anemia grave de origen renal acompañada de síntomas clínicos (ver sección 4.4).
Binocrit está indicado en adultos que reciben quimioterapia por tumores sólidos, linfoma maligno o mieloma múltiple y con riesgo de transfusión, según lo indique el estado general del paciente (situación cardiovascular, anemia preexistente al inicio de la quimioterapia) para el tratamiento de la anemia y reducción. requisitos de transfusión.
Binocrit está indicado en adultos que forman parte de un programa de predonación autóloga para aumentar la producción de sangre autóloga.El tratamiento solo debe realizarse en pacientes con anemia moderada (rango de concentración de hemoglobina [Hb] entre 10 y 13 g / dL [6.2 y 8.1 mmol / L], sin deficiencia de hierro), cuando las técnicas para ahorrar sangre no están disponibles o son insuficientes y planificadas La cirugía electiva mayor requiere una gran cantidad de sangre (4 o más unidades de sangre para las mujeres, 5 o más unidades para los hombres).
Binocrit está indicado en adultos sin deficiencia de hierro, que se cree que tienen un alto riesgo de complicaciones por transfusión, antes de una cirugía ortopédica electiva mayor, para reducir la exposición a transfusiones de sangre alogénica. Limite el uso a pacientes con anemia moderada (rango de concentración de hemoglobina entre 10 y 13 g / dL o entre 6.2 y 8.1 mmol / L) que no forman parte de un programa de predonación autóloga y para el cual se espera una pérdida moderada de sangre (900-1,800 mL).
04.2 Posología y forma de administración -
La terapia con Binocrit debe iniciarse bajo la supervisión de médicos con experiencia en el tratamiento de pacientes con las indicaciones anteriores.
Dosis
Todas las demás causas de anemia (deficiencia de hierro, ácido fólico o vitamina B12, intoxicación por aluminio, infección o inflamación, pérdida de sangre, hemólisis y fibrosis de la médula ósea de cualquier origen) deben evaluarse y tratarse antes de iniciar el tratamiento con epoyetina alfa y al decidir un tratamiento. aumento de dosis. Para garantizar una respuesta óptima a la epoyetina alfa, es necesario asegurarse de que existen reservas de hierro adecuadas y, si es necesario, administrar un suplemento de hierro (ver sección 4.4).
Tratamiento de la anemia sintomática en pacientes adultos con insuficiencia renal crónica
Los síntomas y las consecuencias de la anemia pueden variar según la edad, el sexo y las comorbilidades médicas; el médico requiere una evaluación individual del curso clínico y la condición de cada paciente individual.
El rango recomendado de concentración de hemoglobina deseada es de 10 g / dL a 12 g / dL (6.2 a 7.5 mmol / L). Se debe administrar Binocrit para que los valores de hemoglobina no aumenten más allá de 12 g / dL (7.5 mmol / L). Debe evitarse un aumento de hemoglobina superior a 2 g / dL (1,25 mmol / L) durante un período de semanas. Si esto ocurre, se debe realizar un ajuste de dosis.
Debido a la variabilidad intrapaciente, ocasionalmente se pueden observar valores de hemoglobina individuales por encima y por debajo del rango de concentración de hemoglobina deseado: 10 g / dL (6,2 mmol / L) y 12 g / dL (7,5 mmol / L).
Deben evitarse niveles prolongados de hemoglobina superiores a 12 g / dL (7,5 mmol / L). Si la hemoglobina aumenta en más de 2 g / dL (1,25 mmol / L) por mes, o si tiene niveles de hemoglobina sostenidos por encima de 12 g / dL (7,5 mmol / L), reduzca la dosis de Binocrit en un 25%. Si la hemoglobina excede 13 g / dL (8,1 mmol / L), suspender el tratamiento hasta que los valores caigan por debajo de 12 g / dL (7,5 mmol / L) y luego reanudar el tratamiento con Binocrit a una dosis un 25% menor que la dosis anterior.
Los pacientes deben ser monitoreados de cerca para asegurar que se use la dosis efectiva más baja aprobada de Binocrit para el control adecuado de la anemia y los síntomas de la anemia, mientras se mantiene una concentración de hemoglobina por debajo o igual a 12 g / dL (7.45 mmol / L).
Tenga cuidado al aumentar las dosis de Binocrit en pacientes con insuficiencia renal crónica En pacientes con mala respuesta de hemoglobina a Binocrit, se deben considerar explicaciones alternativas para la mala respuesta (ver secciones 4.4 y 5.1).
El tratamiento con Binocrit consta de dos fases: la fase de corrección y la fase de mantenimiento.
Pacientes adultos en hemodiálisis
En pacientes en hemodiálisis donde el acceso intravenoso está fácilmente disponible, es preferible la administración intravenosa.
Fase de corrección
La dosis inicial es de 50 UI / kg, tres veces por semana.
Si es necesario, aumente o disminuya la dosis en 25 UI / kg (tres veces por semana) hasta alcanzar el rango de concentración de hemoglobina deseado, entre 10 g / dL y 12 g / dL (entre 6.2 y 7.5 mmol / L) (esto debe suceder gradualmente a intervalos de al menos cuatro semanas).
Fase de mantenimiento
La dosis semanal total recomendada está entre 75 UI / kg y 300 UI / kg.
Se debe realizar un ajuste de dosis adecuado para mantener los valores de hemoglobina dentro del rango de concentración deseado de 10 g / dL a 12 g / dL (6.2 a 7.5 mmol / L).
Pacientes con valores de hemoglobina inicialmente muy bajos (8 g / dL o> 5 mmol / L).
Pacientes adultos con insuficiencia renal que aún no reciben diálisis
Cuando el acceso intravenoso no esté fácilmente disponible, Binocrit se puede administrar por vía subcutánea.
Fase de corrección
Dosis inicial de 50 UI / kg, 3 veces por semana, seguida, si es necesario, de incrementos de 25 UI / kg (3 veces por semana) hasta alcanzar el valor deseado (esto debe hacerse gradualmente a intervalos de al menos cuatro semanas ).
Fase de mantenimiento
Durante la fase de mantenimiento, Binocrit se puede administrar 3 veces por semana o, en el caso de administración subcutánea, una vez a la semana o una vez cada 2 semanas.
Se debe realizar un ajuste apropiado de la dosis y los intervalos de dosificación para mantener los valores de hemoglobina en el nivel deseado: hemoglobina entre 10 g / dL y 12 g / dL (6.2 a 7.5 mmol / L). La extensión del intervalo entre dosis puede requerir un aumento de la dosis.
La dosis máxima no debe exceder las 150 UI / kg 3 veces por semana, 240 UI / kg (hasta un máximo de 20.000 UI) una vez a la semana o 480 UI / kg (hasta un máximo de 40.000 UI) una vez cada 2 semanas.
Pacientes adultos sometidos a diálisis peritoneal
Cuando el acceso intravenoso no esté fácilmente disponible, Binocrit se puede administrar por vía subcutánea.
Fase de corrección
La dosis inicial es de 50 UI / kg, dos veces por semana.
Fase de mantenimiento
La dosis de mantenimiento recomendada es entre 25 UI / kg y 50 UI / kg, dos veces por semana, en 2 inyecciones iguales.
Se debe realizar un ajuste de dosis adecuado para mantener los valores de hemoglobina en el nivel deseado, entre 10 g / dL y 12 g / dL (6.2 a 7.5 mmol / L).
Tratamiento de pacientes adultos con anemia inducida por quimioterapia
Los síntomas y las consecuencias de la anemia pueden variar según la edad, el sexo y la gravedad general de la enfermedad; el médico requiere una evaluación individual del curso clínico y la condición de cada paciente individual.
Binocrit debe administrarse a pacientes anémicos (p. Ej., Con una concentración de hemoglobina ≤ 10 g / dl (6,2 mmol / l)).
La dosis inicial es de 150 UI / kg por vía subcutánea, 3 veces por semana.
Alternativamente, Binocrit se puede administrar a una dosis inicial de 450 UI / kg por vía subcutánea una vez a la semana.
Se debe realizar un ajuste de dosis adecuado para mantener los valores de hemoglobina dentro del rango de concentración deseado de 10 g / dL a 12 g / dL (6.2 a 7.5 mmol / L).
Debido a la variabilidad intrapaciente, ocasionalmente se pueden observar concentraciones únicas de hemoglobina por encima y por debajo del rango de concentración de hemoglobina deseado. Se recomienda abordar la variabilidad de la hemoglobina mediante un manejo óptimo de la dosis, teniendo en cuenta el rango de concentración de hemoglobina. 10 g / dL (6.2 mmol / L) a 12 g / dL (7.5 mmol / L) deseadas Deben evitarse concentraciones de hemoglobina prolongadas por encima de 12 g / dL (7.5 mmol / L); pautas para el ajuste de dosis apropiado para concentraciones de hemoglobina por encima de 12 g / dL (7.5 mmol / L) / L) se describen a continuación.
• ¿Si la concentración de hemoglobina ha aumentado al menos 1 g / dL (0,62 mmol / L) o el recuento de reticulocitos ha aumentado? 40.000 células / mcL por encima del valor inicial después de cuatro semanas de tratamiento, se debe mantener una dosis de 150 UI / kg tres veces por semana o 450 UI / kg una vez por semana.
• Si el aumento de la concentración de hemoglobina es
• Si el aumento de la concentración de hemoglobina es
Ajuste de dosis para mantener una concentración de hemoglobina entre 10 g / dL y 12 g / dL (6.2 y 7.5 mmol / L)
Si la concentración de hemoglobina aumenta en más de 2 g / dL (1,25 mmol / L) por mes, o si el nivel de concentración de hemoglobina supera los 12 g / dL (7,5 mmol / L), reduzca la dosis de Binocrit en aproximadamente un 25-50%.
Si el nivel de concentración de hemoglobina supera los 13 g / dL (8,1 mmol / L), suspenda el tratamiento hasta que los valores caigan por debajo de 12 g / dL (7,5 mmol / L) y luego reanude el tratamiento con Binocrit a una dosis un 25% inferior a la dosis anterior.
La pauta posológica recomendada se muestra en la siguiente tabla:
Los pacientes deben ser monitoreados de cerca para asegurar que la dosis más baja aprobada del agente estimulante de la eritropoyesis (agente estimulante de la eritropoyesis, ESA) para un adecuado control de los síntomas de la anemia.
La terapia con epoetina alfa debe continuar hasta un mes después del final de la quimioterapia.
Tratamiento de pacientes quirúrgicos adultos que participan en un programa de predonación autóloga
Los pacientes con anemia leve (hematocrito entre 33 y 39%), que requieran un depósito previo de 4 o más unidades de sangre, deben ser tratados con 600 UI / kg de Binocrit por vía intravenosa, dos veces por semana, durante tres semanas antes de la cirugía. Binocrit debe administrarse después de que se haya completado el procedimiento de donación.
Tratamiento de pacientes adultos programados para cirugía ortopédica electiva mayor
La dosis recomendada es de 600 UI / kg de Binocrit, administrado por vía subcutánea una vez a la semana durante tres semanas (días -21, -14 y -7) antes de la cirugía y el día de la cirugía (día 0).
En los casos en los que, por motivos médicos, sea necesario reducir el tiempo quirúrgico a menos de tres semanas, se deben administrar 300 UI / kg de Binocrit por vía subcutánea durante 10 días consecutivos antes de la cirugía, el día de la cirugía y en los cuatro días inmediatamente siguientes.
Si el nivel de hemoglobina alcanza o supera los 15 g / dL (9,38 mmol / L) en el período preoperatorio, se debe suspender Binocrit y no se deben administrar dosis posteriores.
Población pediátrica
Tratamiento de la anemia sintomática en pacientes con insuficiencia renal crónica en hemodiálisis
Los síntomas y las consecuencias de la anemia pueden variar según la edad, el sexo y las comorbilidades médicas; el médico requiere una evaluación individual del curso clínico y la condición de cada paciente individual.
En pacientes pediátricos, el rango de concentración de hemoglobina recomendado es de 9,5 g / dl a 11 g / dl (5,9 a 6,8 mmol / l). Se debe administrar Binocrit para que los valores de hemoglobina no aumenten por encima de 11 g / dl (6,8 mmol / l). Debe evitarse un aumento de hemoglobina superior a 2 g / dL (1,25 mmol / L) durante un período de cuatro semanas, debiendo realizarse un ajuste de dosis adecuado.
Los pacientes deben ser monitoreados de cerca para asegurar que se use la dosis más baja aprobada de Binocrit para el control adecuado de la anemia y los síntomas de la anemia.
El tratamiento con Binocrit consta de dos fases: la fase de corrección y la fase de mantenimiento.
En pacientes pediátricos en hemodiálisis donde el acceso intravenoso está fácilmente disponible, es preferible la administración intravenosa.
Fase de corrección
La dosis inicial es de 50 UI / kg por vía intravenosa, 3 veces por semana.
Si es necesario, aumente o disminuya la dosis en 25 UI / kg (tres veces por semana) hasta alcanzar el rango de concentración de hemoglobina deseado, entre 9,5 g / dL y 11 g / dL (entre 5,9 y 6,8 mmol / L) ( esto debe hacerse gradualmente a intervalos de al menos cuatro semanas).
Fase de mantenimiento
Se debe realizar un ajuste de dosis apropiado para mantener los niveles de hemoglobina dentro del rango de concentración deseado de 9,5 g / dl a 11 g / dl (5,9 a 6,8 mmol / l).
Por lo general, los niños que pesan menos de 30 kg necesitan dosis de mantenimiento más altas que los niños que pesan más de 30 kg y los adultos.
Pacientes pediátricos con valores de hemoglobina basal muy bajos (6,8 g / dl o> 4,25 mmol / l).
Anemia en pacientes con insuficiencia renal crónica antes de iniciar diálisis o sometidos a diálisis peritoneal
No se ha establecido la seguridad y eficacia de epoyetina alfa en pacientes con insuficiencia renal crónica con anemia antes del inicio de la diálisis o sometidos a diálisis peritoneal. Los datos actualmente disponibles para el uso subcutáneo de epoetina alfa en estas poblaciones se describen en la sección 5.1, pero no se puede hacer una recomendación posológica.
Tratamiento de pacientes pediátricos con anemia inducida por quimioterapia
No se ha establecido la seguridad y eficacia de epoyetina alfa en pacientes pediátricos sometidos a quimioterapia (ver sección 5.1).
Tratamiento de pacientes quirúrgicos pediátricos que participan en un programa de predonación autóloga
No se ha establecido la seguridad y eficacia de epoyetina alfa en pacientes pediátricos No hay datos disponibles.
Tratamiento de pacientes pediátricos en espera de cirugía ortopédica electiva mayor
No se ha establecido la seguridad y eficacia de epoyetina alfa en pacientes pediátricos No hay datos disponibles.
Método de administración
Precauciones que deben tomarse antes de manipular o administrar el medicamento.
Antes de usar, deje reposar la jeringa de Binocrit hasta que alcance la temperatura ambiente, lo que suele tardar entre 15 y 30 minutos.
Al igual que con el resto de productos inyectables, compruebe que la solución no contiene partículas y no presenta decoloración. Binocrit es un producto estéril pero sin conservantes y es para un solo uso. Administrar la cantidad requerida.
Tratamiento de la anemia sintomática en pacientes adultos con insuficiencia renal crónica
En pacientes con insuficiencia renal crónica en los que se dispone regularmente de acceso intravenoso (pacientes en hemodiálisis), es preferible la administración intravenosa de Binocrit.
Cuando el acceso intravenoso no esté fácilmente disponible (pacientes que aún no están en diálisis y pacientes que se someten a diálisis peritoneal), Binocrit puede administrarse mediante inyección subcutánea.
Tratamiento de pacientes adultos con anemia inducida por quimioterapia
Binocrit debe administrarse mediante inyección subcutánea.
Tratamiento de pacientes quirúrgicos adultos que participan en un programa de predonación autóloga
Binocrit debe administrarse por vía intravenosa.
Tratamiento de pacientes adultos programados para cirugía ortopédica electiva mayor
Binocrit debe administrarse mediante inyección subcutánea.
Tratamiento de la anemia sintomática en pacientes pediátricos con insuficiencia renal crónica en hemodiálisis
En pacientes pediátricos con insuficiencia renal crónica en los que se dispone regularmente de acceso intravenoso (pacientes en hemodiálisis), es preferible la administración intravenosa de Binocrit.
Administracion intravenosa
Administrar durante al menos uno a cinco minutos, dependiendo de la dosis total. En pacientes en hemodiálisis, se puede administrar un bolo durante la sesión de diálisis a través de un acceso venoso adecuado en la línea de diálisis. Alternativamente, la inyección se puede administrar al final de la sesión de diálisis a través del tubo de la aguja de la fístula, seguido de 10 ml de solución salina isotónica para enjuagar el tubo y garantizar que el producto se inyecte en la circulación (consulte Posología, "Pacientes adultos en hemodiálisis"). ).
En pacientes que reaccionan al tratamiento con síntomas similares a los de la gripe, es preferible una administración más lenta (ver sección 4.8).
No administrar Binocrit mediante perfusión intravenosa o en combinación con otros medicamentos en solución (consultar la sección 6.6 para obtener más información).
Administración subcutánea
No exceda el volumen máximo de 1 ml en cada sitio de inyección. Para inyectar volúmenes más grandes, use varios sitios de inyección.
Administre la inyección en las extremidades o en la pared abdominal anterior.
En los casos en que el médico determine que el paciente o su cuidador pueden administrar Binocrit por vía subcutánea por sí solos de forma segura y eficaz, se debe proporcionar orientación sobre la dosis y el método de administración correctos.
Las "instrucciones de autoinyección de Binocrit" se encuentran al final del prospecto.
04.3 Contraindicaciones -
• Hipersensibilidad al principio activo oa alguno de los excipientes incluidos en la sección 6.1.
• No administre Binocrit ni ninguna otra eritropoyetina a pacientes que desarrollen aplasia pura de glóbulos rojos (PRCA) después del tratamiento con eritropoyetina (ver sección 4.4).
• Hipertensión incontrolada.
• Todas las contraindicaciones asociadas con los programas de predonación de sangre autóloga deben observarse en pacientes que reciben un suplemento de Binocrit.
El uso de Binocrit en pacientes programados para cirugía ortopédica electiva mayor, que no están participando en un programa de predonación autóloga, está contraindicado en pacientes con enfermedad coronaria, arterial periférica, carotídea o vascular cerebral severa, incluyendo pacientes con infarto de miocardio reciente accidente cerebrovascular o miocárdico. .
• Pacientes quirúrgicos que, por cualquier motivo, no puedan recibir una profilaxis antitrombótica adecuada.
04.4 Advertencias especiales y precauciones de uso apropiadas -
Consideración general
En los pacientes tratados con epoyetina alfa, la presión arterial debe vigilarse y controlarse estrechamente según sea necesario. Epoyetina alfa debe utilizarse con precaución en presencia de hipertensión no tratada, tratada inadecuadamente o mal controlada. Puede ser necesario añadir medicamentos. O aumentar la dosis de terapia antihipertensiva Suspender el tratamiento con epoetina alfa si no se puede controlar la presión arterial.
También se han producido crisis hipertensivas con encefalopatía y convulsiones que requieren atención médica inmediata y cuidados intensivos durante el tratamiento con epoyetina alfa en pacientes con presión arterial previamente normal o baja, similar a la migraña, que puede ser un signo de advertencia (ver sección 4.8).
La epoyetina alfa debe usarse con precaución en pacientes con epilepsia, antecedentes de convulsiones o afecciones médicas asociadas con una predisposición a la actividad convulsiva, como infecciones del SNC y metástasis cerebrales.
La epoetina alfa debe usarse con precaución en pacientes con insuficiencia hepática crónica La seguridad de epoetina alfa no está establecida en pacientes con disfunción hepática.
Se ha observado una mayor incidencia de episodios trombóticos vasculares (TEV) en pacientes tratados con AEE (ver sección 4.8). Estos incluyen trombosis y embolias venosas y arteriales (incluidos algunos casos con desenlace mortal), como trombosis venosa profunda, embolia pulmonar. , trombosis retiniana e infarto de miocardio Además, se han notificado accidentes cerebrovasculares (incluidos infarto cerebral, hemorragia cerebral y ataques isquémicos transitorios).
El riesgo notificado de estos TEV debe sopesarse cuidadosamente con los beneficios esperados del tratamiento con epoyetina alfa, especialmente en pacientes con factores de riesgo preexistentes de TEV, incluida la obesidad y antecedentes de TEV (p. Ej., Trombosis venosa profunda, embolia pulmonar y accidente cerebrovascular). .
En todos los pacientes, los niveles de hemoglobina deben ser monitoreados de cerca debido al riesgo potencialmente mayor de eventos tromboembólicos y desenlace fatal si los pacientes son tratados con niveles de hemoglobina por encima del rango de concentración indicado.
Durante el tratamiento con epoyetina alfa, puede producirse un aumento moderado, dependiente de la dosis, en el recuento de plaquetas dentro del rango normal. Este aumento retrocede durante la continuación del tratamiento. Además, se han notificado casos de trombocitemia por encima del rango normal. Se recomienda. recuento monitoreado regularmente durante las primeras ocho semanas de terapia.
Todas las demás causas de anemia (deficiencia de hierro, ácido fólico o vitamina B12, intoxicación por aluminio, infección o inflamación, pérdida de sangre, hemólisis y fibrosis de la médula ósea de cualquier origen) deben evaluarse y tratarse antes de iniciar el tratamiento con epoyetina alfa y al decidir un tratamiento. aumento de dosis. En la mayoría de los casos, los valores de ferritina sérica disminuyen al mismo tiempo que aumenta el hematocrito. Para garantizar una respuesta óptima a la epoyetina alfa, es necesario asegurarse de que existan reservas de hierro adecuadas y, si es necesario, administrar un suplemento de hierro (ver sección 4.2):
• En pacientes con insuficiencia renal crónica, se recomienda la administración de hierro (hierro elemental, 200 a 300 mg / día por vía oral en adultos y 100 a 200 mg / día por vía oral en sujetos pediátricos) si los niveles séricos de ferritina son inferiores a 100 ng / ml.
• En pacientes con cáncer, se recomienda la administración de hierro (hierro elemental, 200 a 300 mg / día por vía oral) si la saturación de transferrina es inferior al 20%.
• En pacientes que participan en un programa de predonación autóloga, la administración de hierro (hierro elemental, 200 mg / día por vía oral) debe realizarse varias semanas antes de iniciar la predonación autóloga, con el fin de formar abundantes depósitos de hierro antes del "inicio de la terapia con epoetina alfa, y durante todo el tratamiento con epoetina alfa.
• En pacientes programados para cirugía ortopédica electiva mayor, la administración de hierro (hierro elemental, 200 mg / día por vía oral) debe realizarse durante toda la duración del tratamiento con epoetina alfa. Si es posible, la administración de hierro debe iniciarse antes del inicio del tratamiento con epoetina alfa. de modo que se formen reservas de hierro adecuadas.
En muy raras ocasiones, se ha observado la aparición o exacerbación de porfiria en pacientes tratados con epoyetina alfa, que debe utilizarse con precaución en pacientes con porfiria.
Para mejorar la trazabilidad de los agentes estimulantes de la eritropoyesis (AEE), el nombre del agente administrado debe registrarse (o indicarse) de forma inequívoca en la historia clínica del paciente.
Los pacientes deben cambiar de un AEE a otro solo bajo la supervisión adecuada.
Aplasia pura de glóbulos rojos (PRCA)
Se ha informado PRCA mediada por anticuerpos después de meses o años de tratamiento con epoetina subcutánea, principalmente en pacientes con insuficiencia renal crónica. También se han notificado casos en pacientes con hepatitis C tratados con interferón y ribavirina, en presencia de terapia concomitante con AEE. La epoetina alfa no está aprobada para el tratamiento de la anemia asociada con la hepatitis C.
En pacientes en los que de repente hay una falta de eficacia de la terapia, definida por una disminución de la hemoglobina (1-2 g / dL o 0,62-1,25 mmol / L por mes) con un aumento de las necesidades de transfusión, se debe determinar el recuento de reticulocitos y el típico Deben analizarse las causas de la falta de respuesta (deficiencia de hierro, folato o vitamina B12, intoxicación por aluminio, infección o inflamación, pérdida de sangre, hemólisis y fibrosis de la médula ósea de cualquier origen).
En caso de disminución paradójica de la hemoglobina y aparición de anemia grave asociada con recuentos bajos de reticulocitos, se debe suspender el tratamiento con epoetina alfa y determinar los anticuerpos anti-eritropoyetina. También se debe considerar un examen de médula ósea para un "posible diagnóstico de PRCA".
No se deben iniciar otras terapias con AEE debido al riesgo de reacción cruzada.
Tratamiento de la anemia sintomática en pacientes adultos y pediátricos con insuficiencia renal crónica
En pacientes con insuficiencia renal crónica tratados con epoyetina alfa, los niveles de hemoglobina deben medirse regularmente hasta que se alcance un nivel estable y, posteriormente, a intervalos periódicos.
En pacientes con insuficiencia renal crónica, el aumento de hemoglobina debe ser de aproximadamente 1 g / dL (0,62 mmol / L) por mes y no debe exceder los 2 g / dL (1,25 mmol / L) por mes, para minimizar el riesgo de empeoramiento. de hipertensión.
En pacientes con insuficiencia renal crónica, la concentración de hemoglobina de mantenimiento no debe exceder el límite superior del rango de concentración de hemoglobina, como se recomienda en la sección 4.2. Se observó un aumento del riesgo de muerte y eventos cardiovasculares graves en los ensayos clínicos. Nivel de concentración de hemoglobina superior a 12 g / dL (7,5 mmol / L).
Los ensayos clínicos controlados no han demostrado ningún beneficio significativo atribuible a la administración de epoetinas una vez que la concentración de hemoglobina ha superado los niveles necesarios para controlar los síntomas de la anemia y evitar las transfusiones de sangre.
Se debe tener precaución al aumentar las dosis de Binocrit en pacientes con insuficiencia renal crónica, ya que las dosis acumuladas altas de epoetina pueden estar asociadas con un mayor riesgo de mortalidad y eventos cardiovasculares y cerebrovasculares graves. En pacientes con mala respuesta de la hemoglobina a las epoetinas. Explicaciones alternativas para debe tenerse en cuenta la mala respuesta (ver secciones 4.2 y 5.1).
Los pacientes con insuficiencia renal crónica tratados con epoetina alfa subcutánea deben ser controlados periódicamente para detectar la pérdida de eficacia, definida como respuesta nula o reducida al tratamiento con epoetina alfa en pacientes que respondieron previamente a dicha terapia. Esta afección se caracteriza por una disminución prolongada de la hemoglobina a pesar de un aumento de la dosis de epoyetina alfa (ver sección 4.8).
Algunos pacientes que utilizan intervalos más prolongados entre las dosis de epoetina alfa (más de una vez a la semana) pueden no mantener niveles adecuados de hemoglobina (ver sección 5.1) y pueden requerir un aumento de la dosis de epoetina alfa. Los niveles de hemoglobina deben controlarse con regularidad.
Se ha producido trombosis de la derivación en pacientes en hemodiálisis, especialmente en pacientes con tendencia a la hipotensión o con complicaciones de fístulas arteriovenosas (p. Ej., Estenosis, aneurismas, etc.). En estos pacientes, se recomienda la revisión temprana de la derivación y una revisión temprana de la derivación. profilaxis antitrombótica, por ejemplo con ácido acetilsalicílico.
Se ha observado hiperpotasemia en casos aislados, aunque no se ha establecido una relación causal. En pacientes con insuficiencia renal crónica, se deben controlar los electrolitos séricos. En presencia de un nivel de potasio sérico elevado o en aumento, además del tratamiento adecuado de la hiperpotasemia, se debe considerar la posibilidad de interrumpir la administración de epoetina alfa hasta que se corrija el nivel de potasio sérico.
Debido al aumento del hematocrito, a menudo es necesario aumentar la dosis de heparina durante la hemodiálisis durante el tratamiento con epoyetina alfa. Si la heparinización no es óptima es posible que se produzca una oclusión del sistema de diálisis.
Según la información disponible actualmente, la corrección de la anemia con epoyetina alfa en pacientes adultos con insuficiencia renal que aún no se someten a diálisis no acelera la progresión de la insuficiencia renal.
Tratamiento de pacientes con anemia inducida por quimioterapia
En pacientes con cáncer tratados con epoetina alfa, los niveles de hemoglobina deben medirse regularmente hasta que se alcance un nivel estable y, posteriormente, a intervalos periódicos.
Las epoetinas son factores de crecimiento que estimulan principalmente la producción de eritrocitos. Los receptores de eritropoyetina se pueden expresar en la superficie de una variedad de células cancerosas. Al igual que con todos los factores de crecimiento, existe la preocupación de que las epoetinas puedan estimular el crecimiento tumoral. No se puede descartar el papel de los AEE en la progresión tumoral. En la reducción de la supervivencia libre de progresión. En ensayos clínicos controlados, el uso de epoetina alfa y otros AEE se asoció con una reducción del control del tumor locorregional o una reducción de la supervivencia global:
• control locorregional reducido en pacientes con cáncer de cabeza y cuello avanzado tratados con radioterapia, cuando se administra para alcanzar un nivel de concentración de hemoglobina superior a 14 g / dl (8,7 mmol / l),
• una reducción en la supervivencia general y un aumento en las muertes atribuidas a la progresión del tumor a los 4 meses en pacientes con cáncer de mama metastásico tratadas con quimioterapia cuando se administra para alcanzar un rango de concentración de hemoglobina de 12-14 g / dL (7.5- 8.7 mmol / L) ,
• un mayor riesgo de muerte cuando se administra para alcanzar un nivel de concentración de hemoglobina de 12 g / dl (7,5 mmol / l) en pacientes con neoplasias activas, no tratados con quimioterapia ni radioterapia. El uso de AEE no está indicado en esta población de pacientes.
• un aumento del 9% en el riesgo de progresión de la enfermedad (EP) o muerte en el grupo de epoetina alfa y terapia estándar (SOC) y un aumento del riesgo del 15%, que no puede excluirse estadísticamente en pacientes con cáncer de mama metastásico tratadas con quimioterapia cuando se administra para lograr un rango de concentración de hemoglobina de 10-12 g / dL (6.2-7.5 mmol / L).
Con base en lo anterior, en algunas condiciones clínicas la transfusión de sangre debe ser el tratamiento preferido para el manejo de la anemia en pacientes con cáncer. La decisión de tratar con eritropoyetina recombinante debe basarse en una evaluación del balance beneficio-riesgo con la participación del paciente individual y debe tener en cuenta el contexto clínico específico. Los factores a considerar en esta evaluación deben incluir el tipo de cáncer y su estadio, el grado de anemia, la "esperanza de vida", el entorno en el que se trata al paciente y la situación del paciente. preferencias (ver sección 5.1).
En pacientes con cáncer que reciben quimioterapia, se debe tener en cuenta el intervalo de 2-3 semanas entre la administración de AEE y la aparición de eritrocitos inducidos por eritropoyetina al evaluar la idoneidad del tratamiento con epoyetina alfa (pacientes con riesgo de transfusión).
Pacientes quirúrgicos que participan en programas de predonación autóloga
Deben observarse todas las advertencias y precauciones especiales relacionadas con los programas de predonación autóloga; en particular, la sustitución de volumen debe realizarse de forma rutinaria.
Pacientes programados para cirugía ortopédica electiva mayor
Siga siempre las buenas prácticas de manejo de sangre en el período perioperatorio.
Los pacientes programados para cirugía ortopédica mayor electiva deben recibir "profilaxis antitrombótica adecuada, ya que pueden ocurrir eventos trombóticos y vasculares en pacientes quirúrgicos, especialmente en pacientes con enfermedad cardiovascular subyacente. También se debe prestar especial atención. Se debe prestar especial atención a pacientes predispuestos a desarrollar TVP (vena profunda Además, en pacientes con hemoglobina basal> 13 g / dl (> 8,1 mmol / l), no se puede excluir la posibilidad de que el tratamiento con epoyetina alfa pueda estar asociado con un mayor riesgo de episodios trombóticos / vasculares posoperatorios. , la epoyetina alfa no debe usarse en pacientes con hemoglobina basal> 13 g / dl (> 8,1 mmol / l).
Excipientes
Este medicamento contiene menos de 1 mmol (23 mg) de sodio por jeringa precargada, es decir, está esencialmente "exento de sodio".
04.5 Interacciones con otros medicamentos y otras formas de interacción -
No hay evidencia de que el tratamiento con epoetina alfa altere el metabolismo de otros medicamentos.
Los medicamentos que reducen la eritropoyesis pueden reducir la respuesta a la epoetina alfa.
Dado que la ciclosporina se une a los eritrocitos, existe la posibilidad de interacción farmacológica. Si se administra epoyetina alfa concomitantemente con ciclosporina, se deben monitorizar los niveles sanguíneos de ciclosporina y ajustar la dosis de ciclosporina a medida que aumenta el hematocrito.
No hay pruebas que indiquen una "interacción entre la epoetina alfa y el factor estimulante de colonias de granulocitos (G-CSF) o el factor estimulante de colonias de granulocitos y macrófagos (GM-CSF) con respecto a la diferenciación o proliferación hematológica en las muestras de biopsia tumoral". in vitro.
En pacientes adultos con cáncer de mama metastásico, la coadministración subcutánea de 40.000 UI / ml de epoetina alfa con 6 mg / kg de trastuzumab no tuvo ningún efecto sobre la farmacocinética de trastuzumab.
04.6 Embarazo y lactancia -
El embarazo
No existen datos o son limitados sobre el uso de epoetina alfa en mujeres embarazadas Los estudios en animales han mostrado toxicidad para la reproducción (ver sección 5.3).
Por lo tanto, la epoetina alfa solo debe usarse durante el embarazo si el beneficio potencial supera el riesgo potencial para el feto. No se recomienda el uso de epoetina alfa en pacientes quirúrgicas embarazadas que formen parte de un programa de predonación autóloga.
Hora de la comida
Se desconoce si la epoetina alfa exógena se excreta en la leche materna. La epoetina alfa debe usarse con precaución en mujeres en período de lactancia. Se debe decidir si interrumpir la lactancia o interrumpir / abstenerse del tratamiento con epoyetina alfa teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento para la mujer.
No se recomienda el uso de epoetina alfa en pacientes quirúrgicas lactantes que participan en un programa de predonación autóloga.
Fertilidad
No hay estudios para determinar el efecto potencial de la epoetina alfa sobre la fertilidad masculina o femenina.
04.7 Efectos sobre la capacidad para conducir y utilizar máquinas.
No se han realizado estudios sobre la capacidad para conducir y utilizar máquinas La influencia de Binocrit sobre la capacidad para conducir o utilizar máquinas es nula o insignificante.
04.8 Efectos indeseables -
Resumen del perfil de seguridad
La reacción adversa al fármaco más frecuente durante el tratamiento con epoyetina alfa es el aumento de la presión arterial dependiente de la dosis o el empeoramiento de la hipertensión preexistente.Se debe controlar la presión arterial, especialmente al inicio del tratamiento (ver sección 4.4).
Las reacciones adversas más frecuentes observadas en los estudios clínicos con epoetina alfa son las siguientes: diarrea, náuseas, vómitos, pirexia y dolor de cabeza. La enfermedad de tipo gripal puede ocurrir especialmente al inicio del tratamiento.
En estudios con intervalos de dosis prolongados en pacientes adultos con insuficiencia renal y que aún no se someten a diálisis, se han notificado casos de congestión del tracto respiratorio, incluidos eventos de congestión del tracto respiratorio superior, congestión nasal y nasofaringitis.
Se ha observado una "mayor incidencia de acontecimientos trombóticos vasculares (TVE) en pacientes tratados con AEE (ver sección 4.4).
Tabla de reacciones adversas
De un total de 3262 sujetos incluidos en 23 estudios aleatorizados, doble ciego, controlados con placebo o con atención estándar, se evaluó el perfil de seguridad general de epoetina alfa en 1,992 sujetos anémicos.Se incluyeron 228 sujetos con insuficiencia renal crónica tratados. con epoyetina alfa en 4 estudios de insuficiencia renal crónica (2 estudios en prediálisis [N = 131 sujetos expuestos con insuficiencia renal crónica] y 2 en diálisis [N = 97 sujetos expuestos con insuficiencia renal crónica]; 1.404 sujetos con cáncer expuestos en 16 estudios sobre anemia debida a quimioterapia; 147 sujetos expuestos en 2 estudios de donación de sangre autóloga y 213 sujetos expuestos en 1 estudio perioperatorio Las reacciones adversas al fármaco notificadas por ≥ 1% de los sujetos tratados con epoyetina alfa en estos estudios se muestran en la siguiente tabla.
Estimación de frecuencia: muy frecuente (≥ 1/10); frecuentes (≥ 1/100,
1 Identificado en la experiencia poscomercialización y categoría de frecuencia estimada según las tasas de notificación espontánea
² Común en diálisis
³ Incluye eventos arteriales y venosos, tanto fatales como no fatales, como trombosis venosa profunda, émbolos pulmonares, trombosis retiniana, trombosis arterial (incluido infarto de miocardio), accidentes cerebrovasculares (incluido infarto cerebral y hemorragia cerebral), ataques isquémicos transitorios, derivación trombosis (incluido el equipo de diálisis) y trombosis en aneurismas de derivación arteriovenosa
4 Discutido en la subsección siguiente y / o sección 4.4.
Descripción de reacciones adversas seleccionadas
Se han notificado reacciones de hipersensibilidad que incluyen casos de exantema (incluida urticaria), reacciones anafilácticas y edema angioneurótico (ver sección 4.4).
También se han producido crisis hipertensivas con encefalopatía y convulsiones que requieren atención médica inmediata y cuidados intensivos durante el tratamiento con epoyetina alfa en pacientes con presión arterial previamente normal o baja, similar a la migraña, que puede ser un signo de advertencia (ver sección 4.4).
Aplasia pura de glóbulos rojos mediada por anticuerpos (en
Población pediátrica con insuficiencia renal crónica en hemodiálisis
Exposición de pacientes pediátricos con insuficiencia renal crónica en hemodiálisis en ensayos clínicos y en experiencia poscomercialización es limitado. No se notificaron reacciones adversas pediátricas específicas en esta población que no se mencionen en la tabla anterior o que sean incompatibles con la enfermedad subyacente.
Notificación de sospechas de reacciones adversas
La notificación de sospechas de reacciones adversas que se produzcan después de la autorización del medicamento es importante, ya que permite un seguimiento continuo de la relación beneficio / riesgo del medicamento. Se ruega a los profesionales sanitarios que notifiquen cualquier sospecha de reacciones adversas a través del sistema nacional de notificación. "Dirección www. agenziafarmaco.gov.it/it/responsabili.
04.9 Sobredosis -
El margen terapéutico de la epoetina alfa es muy amplio. Una sobredosis de epoetina alfa puede provocar efectos que representen una extensión de los efectos farmacológicos de la hormona. En presencia de niveles de hemoglobina excesivamente altos, se puede utilizar la flebotomía. Se debe recurrir a tratamientos de soporte adicionales. .
05.0 PROPIEDADES FARMACOLÓGICAS -
05.1 "Propiedades farmacodinámicas -
Grupo farmacoterapéutico: antianémicos, eritropoyetina, código ATC: B03XA01.
Binocrit es un medicamento biosimilar. Puede encontrar información más detallada en el sitio web de la Agencia Europea de Medicamentos: http://www.ema.europa.eu.
Mecanismo de acción
La eritropoyetina (EPO) es una hormona glicoproteica producida principalmente por el riñón en respuesta a la hipoxia y es el regulador central de la producción de eritrocitos. "La EPO está involucrada en todas las fases del desarrollo de los eritroides y su efecto principal se expresa a nivel de los precursores eritroides. Después de unirse a su receptor en la superficie celular, la EPO activa las vías de transmisión de señales que interfieren con la" apoptosis y estimula la proliferación de células eritroides.
La EPO humana recombinante (epoetina alfa), expresada en células de ovario de hámster chino, tiene una secuencia de 165 aminoácidos idéntica a la de la EPO urinaria humana; las dos sustancias son indistinguibles de los análisis funcionales. El peso molecular aparente de la eritropoyetina está entre 32.000 y 40.000 daltons.
La eritropoyetina es un factor de crecimiento que estimula principalmente la producción de glóbulos rojos. Los receptores de eritropoyetina se pueden expresar en la superficie de diferentes tipos de células cancerosas.
Efectos farmacodinámicos
Voluntarios sanos
Después de dosis únicas (20.000 a 160.000 UI por vía subcutánea) de epoyetina alfa, se observó una respuesta dependiente de la dosis para los marcadores farmacodinámicos estudiados, incluidos reticulocitos, eritrocitos y hemoglobina. Se observó un perfil de concentración-tiempo definido, con pico y retorno a la línea de base, para detectar cambios en el porcentaje de reticulocitos. Se observó un perfil menos definido para los eritrocitos y la hemoglobina En general, todos los marcadores farmacodinámicos aumentaron en proporción lineal a la dosis y la respuesta máxima se logró a niveles de dosis más altos.
Los estudios farmacodinámicos adicionales analizaron 40.000 UI una vez a la semana en comparación con 150 UI / kg 3 veces por semana. A pesar de las diferencias en los perfiles de concentración-tiempo, la respuesta farmacodinámica (medida por los cambios en el porcentaje de reticulocitos, hemoglobina y eritrocitos totales) fue similar entre estos regímenes terapéuticos. En estudios adicionales, se comparó el régimen de 40.000 UI de epoyetina alfa una vez a la semana con dosis que oscilan entre 80.000 y 120.000 UI administradas por vía subcutánea cada dos semanas. En general, según los resultados de estos estudios farmacodinámicos en sujetos sanos, el régimen de 40.000 UI una vez por semana parece ser más eficaz en términos de producción de eritrocitos en comparación con los regímenes quincenales, aunque se observó una producción similar de reticulocitos en los regímenes de una vez por semana y cada dos semanas.
Falla renal cronica
Se ha demostrado que la epoyetina alfa estimula la eritropoyesis en pacientes anémicos con insuficiencia renal crónica, incluidos los pacientes en diálisis y prediálisis. La primera respuesta notable a la epoyetina alfa es un aumento en el recuento de reticulocitos dentro de los 10 días, seguido de un aumento en el recuento de eritrocitos, hemoglobina y hematocrito, generalmente dentro de 2 a 6 semanas. La respuesta de hemoglobina varía de un paciente a otro y puede ser influenciado por los depósitos de hierro y la presencia de enfermedades concomitantes.
Anemia inducida por quimioterapia
Se ha demostrado que la epoyetina alfa, administrada 3 veces por semana o una vez por semana, aumenta la hemoglobina y reduce la necesidad de transfusiones después del primer mes de terapia en pacientes con cáncer anémicos sometidos a quimioterapia.
En un estudio para comparar el régimen terapéutico con 150 UI / kg 3 veces por semana y el régimen terapéutico con 40.000 UI una vez a la semana en sujetos sanos y en sujetos con cáncer anémico, los perfiles temporales de cambios en el porcentaje de reticulocitos, hemoglobina y total Los eritrocitos fueron similares en ambos regímenes tanto en sujetos sanos como en sujetos con cáncer anémico. Las AUC de los respectivos parámetros farmacodinámicos fueron similares en el régimen de 150 UI / kg 3 veces por semana y el régimen de 40.000 UI. una vez a la semana en sujetos sanos y también en sujetos con cáncer anémico.
Pacientes quirúrgicos adultos que participan en un programa de predonación autóloga
Se ha demostrado que la epoyetina alfa estimula la producción de eritrocitos, lo que permite una mayor recolección de sangre autóloga y limita la reducción de la hemoglobina en pacientes adultos que esperan una cirugía mayor electiva, para quienes se cree que el predepósito no satisface completamente la necesidad perioperatoria de sangre. Los efectos más evidentes se observan en pacientes con valores de hemoglobina bajos (≤ 13 g / dl).
Tratamiento de pacientes adultos programados para cirugía ortopédica electiva mayor
En pacientes programados para cirugía ortopédica mayor electiva con valores de hemoglobina pretratamiento> 10 y ≤ 13 g / dL, se ha demostrado que la epoetina alfa reduce el riesgo de recibir transfusiones alogénicas y acelera la recuperación de eritroides (niveles elevados de hemoglobina, niveles de hematocrito y recuentos de reticulocitos).
Eficacia clínica y seguridad
Falla renal cronica
La epoyetina alfa se ha investigado en ensayos clínicos en pacientes adultos anémicos con insuficiencia renal crónica, incluidos pacientes en hemodiálisis y prediálisis, para el tratamiento de la anemia y el mantenimiento del hematocrito en un rango de concentración objetivo del 30 y el 36%.
En los ensayos clínicos con dosis iniciales de 50 a 150 UI / kg tres veces por semana, aproximadamente el 95% de los pacientes respondieron con un aumento clínicamente significativo del hematocrito. Después de aproximadamente dos meses de tratamiento, casi todos los pacientes eran independientes de las transfusiones. Se alcanzó el hematocrito objetivo, la dosis de mantenimiento se estableció individualmente para cada paciente.
En los tres ensayos clínicos más grandes realizados en pacientes adultos en diálisis, la mediana de la dosis de mantenimiento necesaria para mantener el hematocrito entre el 30 y el 36% fue de aproximadamente 75 UI / kg administrada 3 veces por semana.
En un estudio multicéntrico, doble ciego, controlado con placebo, de calidad de vida en pacientes con insuficiencia renal crónica en hemodiálisis, hubo una mejoría clínica y estadísticamente significativa en los pacientes tratados con epoyetina alfa en comparación con placebo en términos de fatiga, síntomas físicos, relaciones y depresión (Cuestionario sobre la enfermedad renal) después de seis meses de terapia. Los pacientes del grupo de epoetina alfa también se inscribieron en un estudio de extensión de etiqueta abierta en el que se demostraron mejoras en la calidad de vida y se mantuvieron durante 12 meses más.
Pacientes adultos con insuficiencia renal que aún no reciben diálisis
En estudios clínicos llevados a cabo en pacientes con insuficiencia renal crónica no en diálisis tratados con epoetina alfa, la duración media del tratamiento fue de casi cinco meses. Estos pacientes respondieron al tratamiento con epoetina alfa de forma similar a la observada en los pacientes en diálisis. En pacientes con insuficiencia renal crónica que no estaban en diálisis, se observó un aumento prolongado y dependiente de la dosis del hematocrito después de la administración intravenosa o subcutánea de epoetina alfa. Las tasas de aumento del hematocrito fueron similares con ambas vías de administración de epoyetina alfa. Además, las dosis de epoetina alfa Se ha demostrado que la epoyetina alfa entre 75 y 150 UI / kg por semana mantiene el hematocrito en valores entre 36 y 38% hasta por seis meses.
En dos estudios con intervalos de dosificación prolongados para epoyetina alfa (3 veces por semana, una vez por semana, una vez cada 2 semanas y una vez cada 4 semanas), algunos pacientes con intervalos de dosificación más prolongados no mantuvieron niveles adecuados de hemoglobina y cumplieron los criterios de retirada del protocolo para hemoglobina. (0% en el grupo de una vez por semana, 3,7% en el grupo de una vez cada 2 semanas y 3,3% en el grupo de una vez cada 4 semanas).
En un estudio prospectivo aleatorizado se evaluaron 1.432 pacientes anémicos con insuficiencia renal crónica que no estaban dializados. Los pacientes fueron asignados al tratamiento con epoetina alfa para mantener un nivel de hemoglobina de 13,5 g / dL (por encima del nivel de concentración de hemoglobina recomendado) o 11,3 g / dL. Un evento cardiovascular importante (muerte, infarto de miocardio, accidente cerebrovascular u hospitalización por insuficiencia cardíaca congestiva) ocurrió en 125 (18%) de los 715 pacientes del grupo con niveles de hemoglobina más altos en comparación con 97 (14%) de los 717 pacientes en el grupo. grupo con niveles más bajos de hemoglobina (índice de riesgo [HR] 1,3; IC del 95%: 1,0, 1,7; p = 0,03).
Se realizaron análisis combinados post-hoc de estudios clínicos de AEE en pacientes con insuficiencia renal crónica (en diálisis, no en diálisis, diabéticos y no diabéticos). Se observó una tendencia a un aumento en el riesgo estimado de mortalidad por todas las causas y eventos cardiovasculares y cerebrovasculares asociados con dosis acumuladas más altas de AEE, independientemente de la presencia o ausencia de diabetes o diálisis (ver sección 4.2 y sección 4.4).
Tratamiento de pacientes con anemia inducida por quimioterapia
La epoyetina alfa se ha investigado en estudios clínicos en pacientes con cáncer anémico con tumores linfoides y sólidos y en pacientes sometidos a diversos regímenes de quimioterapia, incluidos los regímenes de platino y sin platino. En estos estudios se demostró que la epoyetina alfa, administrada 3 veces por semana y una vez una semana, aumenta la hemoglobina y reduce la necesidad de transfusiones después del primer mes de terapia en pacientes con cáncer anémico. En algunos estudios, la fase doble ciego fue seguida por una fase abierta durante la cual todos los pacientes recibieron epoetina alfa y mantenimiento de se observó el efecto.
La evidencia disponible indica que los pacientes con neoplasias hematológicas y tumores sólidos responden de manera equivalente a la terapia con epoetina alfa y que los pacientes con o sin infiltración tumoral de la médula ósea responden de manera equivalente a la terapia con epoetina alfa. La intensidad similar de la quimioterapia en los grupos de epoetina alfa y placebo en los estudios de quimioterapia se demostró por un área similar bajo la curva de tiempo de neutrófilos en pacientes tratados con epoetina alfa y en pacientes tratados con placebo, así como por una proporción similar de pacientes de los grupos de epoetina alfa y placebo cuyos recuentos absolutos de neutrófilos fueron inferiores a 1.000 y 500 células / mcL.
En un estudio prospectivo, aleatorizado, doble ciego y controlado con placebo realizado con 375 pacientes anémicos con diversas neoplasias malignas no mieloides y tratados con quimioterapia sin platino, se observó una reducción significativa de las secuelas relacionadas con la anemia (fatiga). energía y actividad reducida) basado en las siguientes medidas instrumentales y escalas de calificación: Escala de Evaluación Funcional General de la Terapia del Cáncer-Anemia (FACT-An), escala de fatiga FACT-An y Escala Analógica Lineal del Cáncer (CLAS). En estudios controlados, no se observó una mejora significativa en los parámetros de calidad de vida en las escalas EORTC-QLQ-C30 o CLAS, respectivamente.
La supervivencia y la progresión del tumor se analizaron en cinco grandes estudios controlados en los que participaron un total de 2.833 pacientes, incluidos cuatro estudios doble ciego controlados con placebo y un estudio abierto. Estos estudios incluyeron pacientes en quimioterapia (dos estudios) o poblaciones de pacientes en las que los AEE no están indicados: pacientes con cáncer con anemia que no reciben quimioterapia y pacientes con cáncer de cabeza y cuello que reciben radioterapia. En dos estudios, el nivel de concentración de hemoglobina deseado fue> 13 g / dL (8.1 mmol / L); en los estudios restantes osciló entre 12 y 14 g / dl (7,5 a 8,7 mmol / l). En el estudio de etiqueta abierta, no hubo diferencias en la supervivencia general de los pacientes tratados con eritropoyetina humana recombinante y los controles.En los cuatro estudios controlados con placebo, la razón de riesgo de supervivencia general osciló entre 1,25 y 2,47, a favor de los controles. En comparación con los controles, estos estudios observaron un aumento estadísticamente significativo, constante e inexplicable de la mortalidad en pacientes con anemia asociada a varias neoplasias malignas comunes y tratados con eritropoyetina humana recombinante. El resultado de supervivencia global no se explicó suficientemente por las diferencias en la incidencia de trombosis y complicaciones asociadas en sujetos tratados con eritropoyetina humana recombinante y en sujetos de control.
También se realizó un "análisis de datos de pacientes individuales en más de 13.900 pacientes con cáncer (que recibían quimioterapia, radioterapia, quimiorradioterapia o no recibían ningún tratamiento) que participaban en 53 ensayos clínicos controlados con diferentes epoetinas. El metanálisis de los pacientes. Supervivencia global Los datos proporcionaron una estimación puntual de la tasa de riesgo (cociente de riesgo) de 1,06 a favor de los controles (IC del 95%: 1,00; 1,12; 53 estudios y 13,933 pacientes) y para los pacientes con cáncer tratados con quimioterapia, la razón de riesgo para la supervivencia global fue de 1,04 (IC del 95%: 0,97, 1,11; 38 estudios y 10,441 pacientes). Los metanálisis también han demostrado de forma sistemática un riesgo relativo significativamente mayor de acontecimientos tromboembólicos en pacientes con cáncer tratados con eritropoyetina humana recombinante (ver sección 4.4).
Se realizó un estudio multicéntrico, aleatorizado y abierto en 2098 mujeres con anemia con cáncer de mama metastásico que recibieron quimioterapia de primera o segunda línea. Este fue un estudio de no inferioridad diseñado para excluir un aumento del 15% en el riesgo de progresión tumoral o muerte con epoetina alfa más terapia estándar (SOC) en comparación con SOC solo. Supervivencia libre de progresión mediana (supervivencia libre de progresión, SLP) según la evaluación del investigador de la progresión de la enfermedad fue de 7,4 meses en cada brazo (HR 1,09, IC del 95%: 0,99, 1,20), lo que indica que no se alcanzó el objetivo del estudio. cortar Se reportaron 1337 muertes. La mediana de supervivencia global en el grupo que recibió epoetina alfa más SOC fue de 17,2 meses en comparación con 17,4 meses en el grupo que recibió SOC solo (HR 1,06, IC del 95%: 0,95, 1,18). En el brazo que recibió epoetina alfa más SOC, significativamente menos pacientes recibieron transfusiones de eritrocitos (5,8% frente a 11,4%); sin embargo, en el brazo que recibió epoetina alfa más SOC, significativamente más pacientes (2,8% frente a 1,4%) experimentaron episodios vasculares trombóticos.
Programa de predonación autóloga
El efecto de la epoyetina alfa sobre la facilitación de la donación de sangre autóloga en pacientes con bajo hematocrito (≤ 39% en ausencia de anemia ferropénica subyacente) en espera de una cirugía ortopédica mayor se evaluó en un estudio doble ciego controlado con placebo realizado en 204 pacientes y un estudio simple ciego controlado con placebo realizado en 55 pacientes.
En el estudio doble ciego, los pacientes fueron tratados con epoyetina alfa 600 UI / kg o placebo por vía intravenosa una vez al día cada 3 o 4 días durante 3 semanas (para un total de 6 dosis). En promedio, los pacientes tratados con epoetina alfa pudieron donar significativamente más unidades de sangre para el predepósito (4,5 unidades) en comparación con los pacientes tratados con placebo (3,0 unidades).
En el estudio simple ciego, los pacientes fueron tratados con epoyetina alfa 300 UI / kg o 600 UI / kg o placebo por vía intravenosa una vez al día cada 3 o 4 días durante 3 semanas (para un total de 6 dosis). Estos pacientes tratados con epoetina alfa también pudieron donar significativamente más unidades de sangre al predepósito (epoetina alfa 300 UI / kg = 4,4 unidades; epoetina alfa 600 UI / kg = 4,7 unidades) en comparación con los pacientes tratados con placebo (2,9 unidades ).
La terapia con epoetina alfa redujo el riesgo de exposición a sangre alogénica en un 50% en comparación con los pacientes que no recibieron epoetina alfa.
Cirugía ortopédica electiva mayor
El efecto de la epoyetina alfa (300 UI / kg o 100 UI / kg) sobre la exposición a transfusiones de sangre alogénicas se evaluó en un ensayo clínico doble ciego controlado con placebo en pacientes adultos no ironopénicos en espera de una cirugía ortopédica mayor electiva en la cadera o la rodilla. La epoyetina alfa se administró por vía subcutánea dentro de los 10 días anteriores a la cirugía, el día de la cirugía y durante los cuatro días posteriores a la cirugía. Los pacientes se estratificaron por hemoglobina basal (≤10 g / dL,> 10 a ≤13 g / dL y> 13 g / dL).
Epoyetina alfa 300 UI / kg redujo significativamente el riesgo de transfusión alogénica en pacientes con hemoglobina pretratamiento de> 10 a ≤13 g / dL. Dieciséis por ciento de los pacientes tratados con epoetina alfa 300 UI / kg, 23% de los pacientes tratados con epoetina alfa 100 UI / kg y el 45% de los pacientes tratados con placebo requirieron transfusiones.
En un estudio abierto de grupos paralelos en sujetos adultos sin deficiencia de hierro con hemoglobina pretratamiento de ≥10 a ≤13 g / dl en espera de una cirugía ortopédica mayor de cadera o rodilla, se comparó epoyetina alfa.300 UI / kg por día por vía subcutánea en los 10 días previos a la cirugía, el día de la cirugía y en los cuatro días posteriores a la cirugía con epoetina alfa 600 UI / kg por vía subcutánea una vez a la semana durante las 3 semanas previas a la intervención y el día de la intervención.
Desde la fase de pretratamiento hasta la fase preoperatoria, el aumento medio de hemoglobina en el grupo de 600 UI / kg por semana (1,44 g / dL) fue el doble que en el grupo de 300 UI / dL. Kg por día (0,73 g / dL). Los niveles medios de hemoglobina fueron similares en los dos grupos de tratamiento durante el período posoperatorio.
La respuesta eritropoyética observada en ambos grupos de tratamiento resultó en tasas de transfusión similares (16% en el grupo de 600 UI / kg por semana y 20% en el grupo de 300 UI / kg por día).
Población pediátrica
Falla renal cronica
La epoyetina alfa se evaluó en un ensayo clínico abierto, no aleatorizado, de rango abierto, de 52 semanas en pacientes pediátricos con insuficiencia renal crónica sometidos a hemodiálisis. La mediana de edad de los pacientes incluidos en el estudio fue de 11,6 años (rango de 0,5 a 20,1 años). años).
La epoyetina alfa se administró a dosis de 75 UI / kg / semana por vía intravenosa, divididas en 2 o 3 dosis después de la diálisis, tituladas a 75 UI / kg / semana a intervalos de 4 semanas (hasta un máximo de 300 UI / kg / semana). para obtener un aumento de hemoglobina de 1 g / dL / mes. El rango de concentración de hemoglobina deseado era de 9,6 a 11,2 g / dL. El 81% de los pacientes alcanzó este nivel de concentración de hemoglobina. La mediana del tiempo hasta el objetivo fue de 11 semanas y la dosis media hasta el objetivo fue de 150 UI / kg / semana. De los pacientes que lograron el objetivo, el 90% lo logró con el régimen de 3 veces por semana.
Después de 52 semanas, el 57% de los pacientes permanecieron en el estudio, recibiendo una dosis media de 200 UI / kg / semana.
Los datos clínicos relacionados con la administración subcutánea en niños son limitados. En 5 estudios abiertos no controlados con un número reducido de pacientes (el número de pacientes varió de 9 a 22, para un total de N = 72), se administró epoyetina alfa por vía subcutánea a niños con una dosis única a partir de 100 UI / kg. / semana a 150 UI / kg / semana, con posibilidad de incrementarlo hasta 300 UI / kg / semana. En estos estudios, la mayoría de los pacientes fueron prediálisis (N = 44), 27 pacientes en diálisis peritoneal y dos estaban en hemodiálisis, la edad de los pacientes osciló entre 4 meses y 17 años. En general, estos estudios tienen limitaciones metodológicas, pero el tratamiento se ha asociado con tendencias positivas hacia niveles más altos de hemoglobina. No se informaron eventos adversos inesperados (ver sección 4.2).
Anemia inducida por quimioterapia
Epoyetina alfa 600 UI / kg (administrada por vía intravenosa o subcutánea una vez a la semana) se evaluó en un estudio aleatorizado, doble ciego, controlado con placebo de 16 semanas y en un ensayo aleatorizado, controlado, abierto y con una duración de 20 semanas en pacientes pediátricos con anemia sometida a quimioterapia mielosupresora para el tratamiento de diversas neoplasias no mieloides infantiles.
En el estudio de 16 semanas (n = 222), en pacientes tratados con epoyetina alfa no hubo efectos significativos sobre la calidad de vida en pacientes pediátricos informados por los propios pacientes o por sus padres o en las puntuaciones del Módulo de cáncer en comparación con placebo ( criterio de valoración principal de eficacia) Además, no hubo diferencia estadística entre el porcentaje de pacientes que requirieron transfusiones de glóbulos rojos en el grupo que recibió epoetina alfa y el que recibió placebo.
En el estudio de 20 semanas (n = 225), no hubo diferencias significativas en la variable principal de eficacia, es decir, en la proporción de pacientes que requirieron transfusión de glóbulos rojos después del día 28 (62% de los pacientes que recibieron epoetina alfa frente a 69 % de pacientes que reciben terapia estándar).
05.2 "Propiedades farmacocinéticas -
Absorción
Después de la inyección subcutánea, los niveles séricos de epoetina alfa alcanzaron su punto máximo entre 12 y 18 horas después de la dosis. No hubo acumulación después de la administración de dosis múltiples de 600 UI / kg por vía subcutánea una vez a la semana.
La biodisponibilidad absoluta de epoetina alfa inyectable subcutánea es aproximadamente del 20% en sujetos sanos.
Distribución
El volumen medio de distribución fue de 49,3 ml / kg tras dosis intravenosas de 50 y 100 UI / kg en sujetos sanos. Después de la administración intravenosa de epoyetina alfa en sujetos con insuficiencia renal crónica, el volumen de distribución varió de 57 a 107 ml / kg después de dosis únicas (12 UI / kg) y de 42 a 64 ml / kg después de dosis múltiples, respectivamente. (48 -192 UI / kg). Por tanto, el volumen de distribución es ligeramente mayor que el espacio plasmático.
Eliminación
La vida media de la epoetina alfa después de la administración intravenosa de dosis múltiples es de aproximadamente 4 horas en sujetos sanos.
Se estima que la vida media después de la administración subcutánea es de aproximadamente 24 horas en sujetos sanos.
El CL / F medio para los regímenes de 150 UI / kg 3 veces por semana y 40 000 UI una vez a la semana en sujetos sanos fue de 31,2 y 12,6 ml / h / kg, respectivamente. El CL / F medio para los regímenes de 150 UI / kg 3 veces por semana y 40 000 UI una vez a la semana en sujetos con cáncer anémico fue de 45,8 y 11,3 ml / h / kg, respectivamente. En la mayoría de los sujetos con cáncer anémico sometidos a quimioterapia cíclica, el CL / F fue menor después de dosis subcutáneas de 40.000 UI una vez a la semana y 150 UI / kg 3 veces por semana en comparación con los valores observados en sujetos sanos.
Linealidad / No linealidad
En sujetos sanos, se observó un aumento proporcional a la dosis en las concentraciones séricas de epoetina alfa después de la administración intravenosa de 150 y 300 UI / kg 3 veces por semana. La administración de dosis únicas entre 300 y 2.400 UI / kg de epoetina alfa por vía subcutánea dio como resultado una correlación lineal entre la Cmax media y la dosis y entre el AUC y la dosis media. Se observó una correlación inversa entre el aclaramiento aparente y la dosis en sujetos sanos.
En estudios para examinar el alargamiento del intervalo de dosificación (40.000 UI una vez a la semana y 80.000, 100.000 y 120.000 UI cada dos semanas), se observó una correlación lineal, pero no proporcional a la dosis, entre la Cmax media y la dosis y entre el AUC medio. y dosis en estado estacionario.
Relaciones farmacocinéticas / farmacodinámicas
La epoetina alfa presenta un efecto relacionado con la dosis sobre los parámetros hematológicos que es independiente de la vía de administración.
Población pediátrica
Se ha observado una semivida de aproximadamente 6,2 a 8,7 horas en pacientes pediátricos con insuficiencia renal crónica tras la administración intravenosa de dosis múltiples de epoetina alfa El perfil farmacocinético de epoetina alfa en niños y adolescentes parece similar al de los adultos.
Los datos farmacocinéticos en recién nacidos son limitados.
Un estudio en 7 bebés prematuros con muy bajo peso al nacer y 10 adultos sanos que recibieron eritropoyetina i.v. sugirió que el volumen de distribución era aproximadamente de 1,5 a 2 veces mayor en los lactantes prematuros que en los adultos sanos y que el aclaramiento era aproximadamente 3 veces mayor en los lactantes prematuros que en los adultos sanos.
Insuficiencia renal
En pacientes con insuficiencia renal crónica, la vida media de la epoetina alfa administrada por vía intravenosa es ligeramente más prolongada, aproximadamente 5 horas, en comparación con sujetos sanos.
05.3 Datos preclínicos sobre seguridad -
En estudios de toxicología a dosis repetidas en perros y ratas, pero no en monos, la terapia con epoetina alfa se ha asociado con fibrosis subclínica de la médula ósea. La fibrosis de la médula ósea es una complicación conocida de la insuficiencia renal crónica en humanos y puede estar relacionada con el hiperparatiroidismo secundario o factores desconocidos. La incidencia de fibrosis de la médula ósea no aumentó en un estudio realizado con pacientes en hemodiálisis tratados durante 3 años con epoetina alfa en comparación con un grupo de control compatible de pacientes en diálisis no tratados con epoetina alfa.
La epoetina alfa no induce mutaciones genéticas en bacterias (prueba de Ames), aberraciones cromosómicas en células de mamíferos, micronúcleos en ratones ni mutaciones genéticas en el locus HGPRT.
No se han realizado estudios de carcinogenicidad a largo plazo. Datos discordantes existentes en la literatura, basados en resultados obtenidos in vitro con muestras de tumores humanos, sugieren un posible papel de las eritropoyetinas en la proliferación tumoral. La importancia clínica es incierta.
En cultivos de células de médula ósea humana, la epoetina alfa estimula específicamente la eritropoyesis y no afecta la leucopoyesis. No hubo efecto citotóxico de la epoetina alfa sobre las células de la médula ósea.
En estudios con animales, se ha demostrado que la epoyetina alfa induce una reducción del peso corporal fetal, un retraso en la osificación y un aumento de la mortalidad fetal cuando se administra en dosis semanales aproximadamente 20 veces la dosis semanal recomendada en humanos. Se desconoce la reducción del aumento de peso corporal y su importancia para los seres humanos a niveles de dosis terapéutica.
06.0 INFORMACIÓN FARMACÉUTICA -
06.1 Excipientes -
Fosfato monobásico de sodio dihidrato
Fosfato disódico dihidrato
Cloruro de sodio
Glicina
Polisorbato 80
Agua para preparaciones inyectables
Ácido clorhídrico (para ajustar el pH)
Hidróxido de sodio (para ajustar el pH)
06.2 Incompatibilidad "-
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
06.3 Período de validez "-
2 años
06.4 Precauciones especiales de conservación
Conservar y transportar refrigerado (entre 2 ° C y 8 ° C). Este rango de temperatura debe observarse estrictamente hasta la administración al paciente.
Para uso ambulatorio, el medicamento se puede sacar de la nevera, sin volver a colocarlo, por un período máximo de 3 días a una temperatura no superior a 25 ° C.Si el medicamento no se ha utilizado al final de este período, debe ser descartado.
No congelar ni agitar.
Conservar en el paquete original para proteger el medicamento de la luz.
06.5 Naturaleza del envase primario y contenido del envase.
Jeringas precargadas (vidrio tipo I), con o sin dispositivo de seguridad de la aguja, con tapón de émbolo (goma de teflón) selladas en blísteres.
Binocrit 1.000 UI / 0,5 ml solución inyectable en jeringa precargada.
Cada jeringa precargada contiene 0,5 ml de solución inyectable.
Envases de 1 o 6 jeringas.
Binocrit 2000 UI / 1 ml solución inyectable en jeringa precargada.
Cada jeringa precargada contiene 1 ml de solución inyectable.
Envases de 1 o 6 jeringas.
Binocrit 3000 UI / 0,3 ml solución inyectable en jeringa precargada.
Cada jeringa precargada contiene 0,3 ml de solución inyectable.
Envases de 1 o 6 jeringas.
Binocrit 4000 UI / 0,4 ml solución inyectable en jeringa precargada.
Cada jeringa precargada contiene 0,4 ml de solución inyectable.
Envases de 1 o 6 jeringas.
Binocrit 5000 UI / 0,5 ml solución inyectable en jeringa precargada.
Cada jeringa precargada contiene 0,5 ml de solución inyectable.
Envases de 1 o 6 jeringas.
Binocrit 6000 UI / 0,6 ml solución inyectable en jeringa precargada.
Cada jeringa precargada contiene 0,6 ml de solución inyectable.
Envases de 1 o 6 jeringas.
Binocrit 7.000 UI / 0,7 ml solución inyectable en jeringa precargada.
Cada jeringa precargada contiene 0,7 ml de solución inyectable.
Envases de 1 o 6 jeringas.
Binocrit 8.000 UI / 0,8 ml solución inyectable en jeringa precargada.
Cada jeringa precargada contiene 0,8 ml de solución inyectable.
Envases de 1 o 6 jeringas.
Binocrit 9.000 UI / 0,9 ml solución inyectable en jeringa precargada.
Cada jeringa precargada contiene 0,9 ml de solución inyectable.
Envases de 1 o 6 jeringas.
Binocrit 10.000 UI / 1 ml solución inyectable en jeringa precargada.
Cada jeringa precargada contiene 1 ml de solución inyectable.
Envases de 1 o 6 jeringas.
Binocrit 20.000 UI / 0,5 ml solución inyectable en jeringa precargada.
Cada jeringa precargada contiene 0,5 ml de solución inyectable.
Envases de 1, 4 o 6 jeringas.
Binocrit 30.000 UI / 0,75 ml solución inyectable en jeringa precargada.
Cada jeringa precargada contiene 0,75 ml de solución inyectable.
Envases de 1, 4 o 6 jeringas.
Binocrit 40.000 UI / 1 ml solución inyectable en jeringa precargada.
Cada jeringa precargada contiene 1 ml de solución inyectable.
Envases de 1, 4 o 6 jeringas.
Es posible que no se comercialicen todos los tamaños de envases.
06.6 Instrucciones de uso y manipulación -
Binocrit no debe utilizarse y debe desecharse.
• si el líquido tiene color o si ve partículas flotando en él
• si el sello está roto
• si se sabe o se supone que puede haber sido congelado accidentalmente o
• si ha ocurrido una falla en el refrigerador
Las jeringas precargadas están listas para su uso (ver sección 4.2). La jeringa precargada no debe agitarse. Las jeringas están marcadas con graduaciones elevadas; esto permite un uso parcial si es necesario. Cada graduación corresponde a un volumen de 0, 1 ml. El producto es para un solo uso. Extraiga solo una dosis de Binocrit de cada jeringa y deseche cualquier solución innecesaria antes de inyectar.
Uso de la jeringa precargada con protector de seguridad para la aguja
El protector de seguridad de la aguja cubre la aguja después de la inyección y evita que el operador se lastime. El dispositivo no interfiere con el uso normal de la jeringa. Empuje el émbolo lenta y uniformemente hasta que se libere la dosis completa y no se pueda empujar más el émbolo. Mientras continúa presionando el émbolo, retire la jeringa del paciente. El dispositivo de seguridad cubre la aguja tan pronto como se suelta el émbolo.
Uso de la jeringa precargada sin protector de seguridad para la aguja
Administre la dosis de acuerdo con el procedimiento estándar.
Los medicamentos no utilizados y los desechos derivados de este medicamento deben eliminarse de acuerdo con las regulaciones locales.
07.0 TITULAR DE LA "AUTORIZACIÓN DE COMERCIALIZACIÓN" -
Sandoz GmbH
Biochemiestr. 10
A-6250 Kundl
Austria
08.0 NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN -
Binocrit 1.000 UI / 0,5 ml solución inyectable en jeringa precargada.
EU / 1/07/410/001 - 1000 UI / 0,5 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 0,5 ml (2000 UI / ml) 1 jeringa precargada - AIC n. 038190017 / E
EU / 1/07/410/002 - 1000 UI / 0,5 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 0,5 ml (2000 UI / ml) 6 jeringas precargadas - AIC n. 038190029 / E
EU / 1/07/410/027 - 1000 UI / 0,5 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 0,5 ml (2000 UI / ml) 1 jeringa precargada de 0,5 ml con dispositivo de seguridad para aguja - AIC n. 038190272 / E
EU / 1/07/410/028 - 1000 UI / 0,5 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea intravenosa - 0,5 ml (2000 UI / ml) - 6 jeringas precargadas de 0,5 ml con aguja para dispositivo seguridad - AIC N. 038190284 / E
Binocrit 2000 UI / 1 ml solución inyectable en jeringa precargada.
EU / 1/07/410/003 - 2000 UI / 1,0 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 1,0 ml (2000 UI / ml) 1 jeringa precargada - AIC n. 038190031 / E
EU / 1/07/410/004 - 2000 UI / 1,0 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 1,0 ml (2000 UI / ml) 6 jeringas precargadas - AIC n. 038190043 / E
EU / 1/07/410/029 - 2000 UI / 1 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 1 ml (2000 UI / ml) 1 jeringa precargada de 1 ml con dispositivo de seguridad para la aguja - AIC N. 038190296 / E
EU / 1/07/410 / 030-2000 UI / 0,5 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea intravenosa - 1 ml (2000 UI / ml) - 6 jeringas precargadas de 1 ml con aguja de seguridad dispositivo - AIC N. 038190308 / E
Binocrit 3000 UI / 0,3 ml solución inyectable en jeringa precargada.
EU / 1/07/410/005 - 3000 UI / 0,3 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 0,3 ml (10000 UI / ml) 1 jeringa precargada - AIC n. 038190056 / E
EU / 1/07/410/006 - 3000 UI / 0,3 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 0,3 ml (10000 UI / ml) 6 jeringas precargadas - AIC n. 038190068 / E
EU / 1/07/410 / 031- 3000 UI / 0,3 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 0,3 ml (10000 UI / ml) 1 jeringa precargada de 0,3 ml con dispositivo de seguridad por aguja - AIC N. 038190310 / E
EU / 1/07/410/032 - 3000 UI / 0,3 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea intravenosa - 0,3 ml (10000 UI / ml) - 6 jeringas precargadas de 0,3 ml con aguja de seguridad del dispositivo - AIC N. 038190322 / E
Binocrit 4000 UI / 0,4 ml solución inyectable en jeringa precargada.
EU / 1/07/410/007 - 4000 UI / 0,4 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 0,4 ml (10000 UI / ml) 1 jeringa precargada - AIC n. 038190070 / E
EU / 1/07/410/008 - 4000 UI / 0,4 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 0,4 ml (10000 UI / ml) 6 jeringas precargadas - AIC n. 038190082 / E
EU / 1/07/410/033 - 4000 UI / 0,4 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 0,4 ml (10000 UI / ml) 1 jeringa precargada de 0,4 ml con dispositivo de seguridad por aguja - AIC N. 038190334 / E
EU / 1/07/410/034 - 4000 UI / 0,4 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea intravenosa - 0,4 ml (10000 UI / ml) - 6 jeringas precargadas de 0,4 ml con dispositivo de seguridad aguja - AIC N. 038190346 / E
Binocrit 5000 UI / 0,5 ml solución inyectable en jeringa precargada.
EU / 1/07/410/009 - 5000 UI / 0,5 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 0,5 ml (10000 UI / ml) 1 jeringa precargada - AIC n. 038190094 / E
EU / 1/07/410/010 - 5000 UI / 0,5 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 0,5 ml (10000 UI / ml) 6 jeringas precargadas - AIC n. 038190106 / E
EU / 1/07/410/035 - 5000 UI / 0,5 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea intravenosa - 0,5 ml (10000 UI / ml) - 1 jeringa precargada de 0,5 ml con aguja de seguridad del dispositivo - AIC N. 038190359 / E
EU / 1/07/410/036 - 5000 UI / 0,5 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea intravenosa - 0,5 ml (10000 UI / ml) - 6 jeringas precargadas de 0,5 ml con dispositivo de seguridad para aguja -AIC N. 038190361 / E
Binocrit 6000 UI / 0,6 ml solución inyectable en jeringa precargada.
EU / 1/07/410/011 - 6000 UI / 0,6 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 0,6 ml (10,000 UI / ml) 1 jeringa precargada - AIC n . 038190118 / E
EU / 1/07/410/012 - 6000 UI / 0,6 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 0,6 ml (10,000 UI / ml) 6 jeringas precargadas - AIC n . 038190120 / E
EU / 1/07/410/037 - 6000 UI / 0,6 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea intravenosa - 0,6 ml (10000 UI / ml) - 1 jeringa precargada de 0,6 ml con dispositivo de seguridad aguja - AIC N. 038190373 / E
EU / 1/07/410/038 - 6000 UI / 0,6 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea intravenosa - 0,6 ml (10,000 UI / ml) - 6 jeringas precargadas de 0 , 6 ml con aguja de seguridad del dispositivo - AIC N. 038190385 / E
Binocrit 7.000 UI / 0,7 ml solución inyectable en jeringa precargada.
EU / 1/07/410/017 - 7000 UI / 0,7 ml solución inyectable en jeringa precargada - vía subcutánea o intravenosa - jeringa precargada (vidrio) 0,7 ml (10000 UI / ml) 1 jeringa precargada - AIC No 038190171 / Y
EU / 1/07/410/018 - 7000 UI / 0,7 ml solución inyectable en jeringa precargada - vía subcutánea o intravenosa - jeringa precargada (vidrio) 0,7 ml (10000 UI / ml) 6 jeringas precargadas - AIC No 038190183 / Y
EU / 1/07/410/039 - 7000 UI / 0,7 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 0,7 ml (10000 UI / ml) 1 jeringa precargada de 0,7 ml con dispositivo de seguridad por aguja - AIC N. 038190397 / E
EU / 1/07/410/040 - 7000 UI / 0,7 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 0,7 ml (10000 UI / ml) 6 jeringas precargadas de 0,7 ml con dispositivo de seguridad por aguja - AIC N. 038190409 / E
Binocrit 8.000 UI / 0,8 ml solución inyectable en jeringa precargada.
EU / 1/07/410/013 - 8000 UI / 0,8 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 0,8 ml (10,000 UI / ml) 1 jeringa precargada - AIC n . 038190132 / E
EU / 1/07/410/014 - 8000 UI / 0,8 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 0,8 ml (10,000 UI / ml) 6 jeringas precargadas - AIC n . 038190144 / E
EU / 1/07/410/041 - 8000 UI / 0,8 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea intravenosa - 0,8 ml (10000 UI / ml) - 1 jeringa precargada de 0,8 ml con dispositivo de seguridad aguja - AIC N. 038190411 / E
EU / 1/07/410/042 - 8000 UI / 0,8 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea intravenosa - 0,8 ml (10000 UI / ml) - 6 jeringas precargadas de 0,8 ml con dispositivo de seguridad aguja - AIC N. 038190423 / E
Binocrit 9.000 UI / 0,9 ml solución inyectable en jeringa precargada.
EU / 1/07/410/019 - 9000 UI / 0,9 ml solución inyectable en jeringa precargada - uso subcutáneo o intravenoso jeringa precargada (vidrio) 0,9 ml (10000 UI / ml) 1 jeringa precargada precargada - AIC No. 038190195 / Y
EU / 1/07/410/020 - 9000IU / 0,9 ml solución inyectable en jeringa precargada - vía subcutánea o intravenosa - jeringa precargada (vidrio) 0,9 ml (10000UI / ml) 6 jeringas precargadas - AIC No 038190207 / Y
EU / 1/07/410/043 - 9000 UI / 0,9 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 0,9 ml (10000 UI / ml) 1 jeringa precargada de 0,9 ml con dispositivo de seguridad por aguja - AIC N. 038190435 / E
EU / 1/07/410/044 - 9000 UI / 0,9 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 0,9 ml (10000 UI / ml) 6 jeringas precargadas de 0,9 ml con dispositivo de seguridad por aguja - AIC N. 038190447 / E
Binocrit 10.000 UI / 1 ml solución inyectable en jeringa precargada.
EU / 1/07/410/015 - 10000 UI / 1,0 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 1,0 ml (10000 UI / ml) 1 jeringa precargada - AIC n. 038190157 / E
EU / 1/07/410/016 - 10000 UI / 1,0 ml solución inyectable en jeringa precargada (vidrio) vía subcutánea o intravenosa 1,0 ml (10000 UI / ml) 6 jeringas precargadas - AIC n. 038190169 / E
EU / 1/07/410/045 - 10000 UI / 1 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea intravenosa - 1 ml (10000 UI / ml) - 1 jeringa precargada de 1 ml con dispositivo de seguridad para aguja - AIC N.038190450 / E
EU / 1/07/410/046 - 10000 UI / 1 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea intravenosa - 1 ml (10000 UI / ml) - 6 jeringas precargadas de 1 ml con dispositivo de seguridad para aguja - AIC N. 038190462 / E
Binocrit 20.000 UI / 0,5 ml solución inyectable en jeringa precargada.
EU / 1/07/410/021 - 20.000 UI / 0,5 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa 0,5 ml (40.000 UI / ml) 1 jeringa precargada - AIC n. 038190219 / E
EU / 1/07/410/022 - 20.000 UI / 0,5 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa 0,5 ml (40.000 UI / ml) 6 jeringas precargadas - AIC n. 038190221 / E
EU / 1/07/410/047 - 20000 UI / 0,5 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 0,5 ml (40000 UI / ml) 1 jeringa precargada de 0,5 ml con dispositivo de seguridad por aguja - AIC N. 038190474 / E
EU / 1/07/410/053 - 20.000 UI / 0,5 ML - Solución inyectable en jeringa precargada - Vía subcutánea o intravenosa - Jeringa precargada (vidrio) 0,5 ML (40.000 UI / ML) - 4 precargadas jeringas con dispositivo de seguridad para aguja AIC: 038190536 / E
EU / 1/07/410/048 - 20000 UI / 0,5 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 0,5 ml (40000 UI / ml) 6 jeringas precargadas de 0,5 ml con dispositivo de seguridad por aguja - AIC N. 038190486 / E
Binocrit 30.000 UI / 0,75 ml solución inyectable en jeringa precargada.
EU / 1/07/410/023 - 30.000 UI / 0,75 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa 0,75 ml (40.000 UI / ml) 1 jeringa precargada - AIC n. 038190233 / E
EU / 1/07/410/024 - 30.000 UI / 0,75 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa 0,75 ml (40.000 UI / ml) 6 jeringas precargadas - AIC n. 038190245 / E
EU / 1/07/410/049 - 30000 UI / 0,75 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 0,75 ml (40000 UI / ml) 1 jeringa precargada de 0,75 ml con dispositivo de seguridad para Agosto - AIC N. 038190498 / E
EU / 1/07/410/054 - 30.000 UI / 0,75 ML - Solución inyectable en jeringa precargada - Vía subcutánea o intravenosa - Jeringa precargada (vidrio) 0,75 ML (40.000 UI / ML) - 4 precargadas jeringas con dispositivo de seguridad para aguja AIC: 038190548 / E
EU / 1/07/410/050 - 30000 UI / 0,75 ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 0,75 ml (40000 UI / ml) 6 jeringas precargadas de 0,75 ml con dispositivo de seguridad por aguja - AIC N. 038190500 / E
Binocrit 40.000 UI / 1 ml solución inyectable en jeringa precargada.
EU / 1/07/410/025 - 40.000 UI / 1,0 ml - solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 1,0 ml (40.000 UI / ml) 1 jeringa precargada - AIC n. 038190258 / E
EU / 1/07/410/026 - 40.000 UI / 1,0 ml - solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 1,0 ml (40.000 UI / ml) 6 jeringas precargadas - AIC n. 038190260 / E
EU / 1/07/410/051 - 40000UI / 1ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 1ml (40000IU / ml) 1 jeringa precargada de 1ml con dispositivo de seguridad de aguja - AIC N. 038190512 / E
EU / 1/07/410/055 - 40.000 UI / 1.0 ML - Solución inyectable en jeringa precargada - Vía subcutánea o intravenosa - Jeringa precargada (vidrio) 1.0 ML (40.000 UI / ML) - 4 precargadas jeringas con dispositivo aguja de seguridad para aguja AIC: 038190551 / E
EU / 1/07/410/052 - 40000UI / 1ml solución inyectable en jeringa precargada (vidrio) - vía subcutánea o intravenosa - 1ml (40000IU / ml) 6 jeringas precargadas de 1ml con dispositivo de seguridad para aguja - AIC N. 038190524 / E
09.0 FECHA DE LA PRIMERA AUTORIZACIÓN O RENOVACIÓN DE LA AUTORIZACIÓN -
Desde la primera autorización: 28 de agosto de 2007
Fecha de la renovación más reciente: 18 de junio de 2012
10.0 FECHA DE REVISIÓN DEL TEXTO -
10/2016