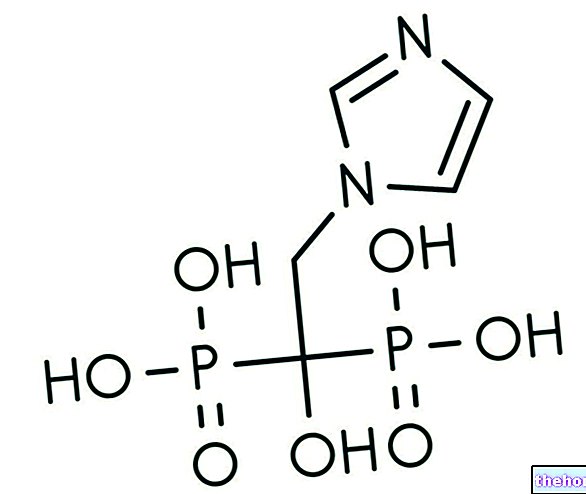
Ingredientes activos: ácido valproico (valproato de sodio)
DEPAKIN 50 mg granulado de liberación modificada
DEPAKIN 100 mg granulado de liberación modificada
DEPAKIN 250 mg granulado de liberación modificada
DEPAKIN 500 mg granulado de liberación modificada
DEPAKIN 750 mg granulado de liberación modificada
DEPAKIN 1000 mg granulado de liberación modificada
Los prospectos de Depakin están disponibles para los siguientes tamaños de envase: - DEPAKIN 50 mg gránulos de liberación modificada, DEPAKIN 100 mg gránulos de liberación modificada, DEPAKIN 250 mg gránulos de liberación modificada, DEPAKIN 500 mg gránulos de liberación modificada, DEPAKIN 750 mg gránulos de liberación modificada, DEPAKIN 1000 mg gránulos de liberación modificada
- DEPAKIN 200 mg comprimidos gastrorresistentes, DEPAKIN 500 mg comprimidos gastrorresistentes, DEPAKIN 200 mg / ml solución oral
- DEPAKIN 400 mg / 4 ml polvo y disolvente para solución para perfusión
¿Por qué se usa Depakin? ¿Para qué sirve?
En el tratamiento de la epilepsia generalizada, en particular en ataques del tipo:
- ausencia
- mioclónico
- tónico
- clónico
- atónico
- mezclado
y en epilepsia parcial:
- simple o complejo
- secundariamente generalizado
En el tratamiento de síndromes específicos (West, Lennox-Gastaut). En el tratamiento de episodios maníacos relacionados con el trastorno bipolar cuando el litio está contraindicado o no se tolera. Se puede considerar la continuación del tratamiento después del episodio de manía en pacientes que han respondido al valproato para la manía aguda.
Contraindicaciones Cuándo no se debe usar Depakin
- Hepatitis aguda
- Hepatitis crónica
- Antecedentes personales o familiares de enfermedad hepática grave, especialmente inducida por fármacos
- Hipersensibilidad al principio activo oa alguno de los excipientes.
- Porfiria hepática
- Trastornos de la coagulación
Precauciones de uso Lo que necesita saber antes de tomar Depakin
En niños menores o iguales a tres años, los antiepilépticos que contienen ácido valproico son solo en casos excepcionales la terapia de primera elección.
- Las pruebas de función hepática deben realizarse antes del inicio de la terapia (ver "Contraindicaciones") y repetirse periódicamente durante los primeros 6 meses, especialmente en pacientes de riesgo (ver "Advertencias especiales").
Como ocurre con la mayoría de los fármacos antiepilépticos, se pueden observar aumentos de las enzimas hepáticas, sobre todo al inicio del tratamiento; son transitorios y aislados, no acompañados de signos clínicos. En estos pacientes, se recomiendan estudios de laboratorio más profundos (incluido el tiempo para la protrombina ), también se puede considerar un ajuste de la dosis y repetir las pruebas si es necesario.
- En niños menores de 3 años, Depakin debe administrarse como monoterapia aunque su beneficio potencial debe evaluarse antes de iniciar el tratamiento, en comparación con el riesgo de daño hepático o pancreatitis en estos pacientes (ver "Advertencias especiales" ").
Se debe evitar el uso concomitante de salicilatos en niños menores de 3 años debido al riesgo de hepatotoxicidad.
- Se recomienda que se realicen análisis de sangre (hemograma completo con recuento de plaquetas, tiempo de sangrado y pruebas de coagulación) antes del inicio del tratamiento o antes de la cirugía y en el caso de hematomas o hemorragias espontáneas (ver "Reacciones adversas").
- En pacientes con insuficiencia renal o hipoproteinemia es necesario disminuir la dosis. Dado que la monitorización de las concentraciones plasmáticas puede ser engañosa, la dosis debe adaptarse de acuerdo con la monitorización clínica.
- Aunque las enfermedades inmunitarias se han encontrado sólo excepcionalmente durante el uso de valproato, vale la pena considerar el beneficio potencial del valproato frente al riesgo potencial en pacientes con lupus eritematoso sistémico.
- Como se han notificado casos excepcionales de pancreatitis, los pacientes con dolor abdominal agudo deben someterse a un examen médico inmediato.En caso de pancreatitis, se debe interrumpir el tratamiento con valproato.
- Si se sospecha un ciclo de urea alterado, se debe evaluar la hiperamonemia antes del tratamiento, ya que es posible que se agrave con el valproato (ver "Reacciones adversas"). Por tanto, si aparecen síntomas como apatía, somnolencia, vómitos, hipotensión y aumento de la frecuencia de convulsiones, se deben determinar los niveles séricos de amoniaco y ácido valproico; si es necesario, se debe reducir la dosis del medicamento. Si se sospecha una interrupción enzimática del ciclo de la urea, se debe determinar el nivel sérico de amoníaco antes de iniciar la terapia con medicamentos que contienen ácido valproico.
- Antes de iniciar el tratamiento, se debe advertir a los pacientes del riesgo de aumento de peso y se deben tomar las medidas adecuadas para minimizar este riesgo (ver "Reacciones adversas").
- Se debe advertir a los pacientes con deficiencia subyacente de carnitina palmitoiltransferasa (CPT) tipo II del aumento del riesgo de rabdomiólisis cuando toman valproato.
- No se recomienda el uso concomitante de ácido valproico / valproato de sodio y medicamentos que contengan carbapenémicos (ver Interacciones).
- Mujeres en edad fértil (ver "Advertencias especiales")
Todas las mujeres con epilepsia y en edad fértil deben estar adecuadamente informadas sobre los riesgos asociados con el embarazo.
- Hematología
Los recuentos de células sanguíneas, incluidos el recuento de plaquetas, el tiempo de hemorragia y las pruebas de coagulación, deben controlarse antes de iniciar el tratamiento, antes de una cirugía o cirugía dental, y en caso de hematomas o hemorragias espontáneas (ver "Reacciones adversas" "). En caso de ingesta concomitante de vitamina Antagonistas de K, se recomienda una estrecha monitorización de los valores de INR.
Interacciones ¿Qué medicamentos o alimentos pueden modificar el efecto de Depakin?
Informe a su médico o farmacéutico si ha tomado recientemente otros medicamentos, incluso sin receta.
Efectos del valproato sobre otras drogas
- Neurolépticos, anti-MAO, antidepresivos y benzodiazepinas
El valproato puede potenciar el efecto de otros fármacos psicotrópicos como neurolépticos, fármacos anti-MAO, antidepresivos y benzodiazepinas, por lo que se recomienda la monitorización clínica y, cuando sea necesario, el ajuste de dosis.
- Fenobarbital
Dado que el valproato aumenta las concentraciones plasmáticas de fenobarbital (por inhibición del catabolismo hepático), puede producirse sedación especialmente en niños. Por tanto, se recomienda la monitorización clínica durante los primeros 15 días de tratamiento combinado, con reducción inmediata de las dosis de fenobarbital en caso de sedación, y posible monitorización de los niveles plasmáticos de fenobarbital.
- Primidona
El valproato aumenta los niveles plasmáticos de primidona potenciando sus efectos indeseables (como la sedación); esta interacción cesa con el tratamiento a largo plazo. Se recomienda la monitorización clínica especialmente al inicio de la terapia combinada con ajuste de la dosis de primidona cuando sea necesario.
- Fenitoína
El valproato inicialmente disminuye la concentración plasmática total de fenitoína pero aumenta su fracción libre, con posibles síntomas de sobredosis (el ácido valproico desplaza la fenitoína de sus sitios de unión a proteínas y ralentiza su catabolismo hepático), por lo que se recomienda la monitorización clínica; en el caso de dosis plasmáticas de fenitoína, debe tenerse en cuenta la fracción libre. Posteriormente, tras el tratamiento crónico, las concentraciones de fenitoína vuelven a los valores iniciales de prevalproato.
- Carbamazepina
Se ha informado toxicidad clínica cuando se administra valproato con carbamazepina, ya que el valproato puede potenciar el efecto tóxico de la carbamazepina. Se recomienda la monitorización clínica especialmente al inicio de la terapia combinada con ajuste de dosis cuando sea necesario.
- Lamotrigina
Depakin reduce el metabolismo de lamotrigina y aumenta su semivida media casi 2 veces. Esta interacción puede conducir a un aumento de la toxicidad de lamotrigina, especialmente erupciones cutáneas graves. Por lo tanto, se recomienda un seguimiento clínico y la dosis de lamotrigina debe reducirse cuando sea necesario.
- Etosuximida
El valproato puede causar un aumento de las concentraciones plasmáticas de etosuximida.
- Zidovudina
El valproato puede aumentar la concentración plasmática de zidovudina dando lugar a un aumento de la toxicidad de la zidovudina.
- Felbamato
El ácido valproico puede disminuir el aclaramiento medio de felbamato hasta en un 16%.
Efectos de otros fármacos sobre el valproato
Los antiepilépticos inductores de enzimas (en particular fenitoína, fenobarbital y carbamazepina) disminuyen las concentraciones séricas de ácido valproico. En caso de terapia combinada, las dosis deben ajustarse de acuerdo con los niveles en sangre.
Por otro lado, la combinación de felbamato y valproato disminuye el aclaramiento de ácido valproico del 22% al 50% y, en consecuencia, aumenta la concentración plasmática de ácido valproico, por lo que debe monitorizarse la tasa plasmática de valproato.
La mefloquina aumenta el metabolismo del ácido valproico y tiene un efecto convulsivo, por lo que pueden ocurrir convulsiones en casos de terapia combinada.
En caso de uso concomitante de valproato y sustancias que se unen en gran medida a proteínas (ácido acetilsalicílico), los niveles séricos libres de ácido valproico pueden aumentar.
Los medicamentos que contienen ácido valproico no deben administrarse concomitantemente con ácido acetilsalicílico para tratar la fiebre y el dolor, especialmente en bebés y niños.
Se debe realizar una estrecha monitorización del tiempo de protrombina en caso de uso concomitante de factores anticoagulantes dependientes de la vitamina K. Los niveles séricos de ácido valproico pueden aumentar (debido a un metabolismo hepático reducido) con el uso concomitante de cimetidina o eritromicina y fluoxetina.
Sin embargo, también se han notificado casos en los que la concentración sérica de ácido valproico se redujo tras la ingesta concomitante de fluoxetina. Cuando se administra concomitantemente con medicamentos que contienen carbapenémicos, se ha informado una disminución de los niveles sanguíneos de ácido valproico, lo que resulta en una reducción del 60-100% en estos niveles sanguíneos en aproximadamente dos días. Debido a su rápida aparición y marcada disminución, la administración concomitante de medicamentos que contienen carbapenémicos en pacientes estabilizados con ácido valproico no se considera factible y, por lo tanto, debe evitarse (ver Precauciones de uso).
La rifampicina puede disminuir los niveles plasmáticos de ácido valproico provocando la interrupción del efecto terapéutico. Por lo tanto, puede ser necesario un ajuste de la dosis de valproato cuando se coadministra con rifampicina.
Otras interacciones
La administración concomitante de valproato y topiramato se ha asociado con la aparición de encefalopatía y / o hiperamonemia.
Los pacientes tratados con estos dos fármacos deben ser controlados con especial atención a los signos y síntomas de encefalopatía hiperamonémica. El valproato generalmente no tiene un efecto inductor de enzimas; en consecuencia, no reduce la eficacia de los estrógenos-progestágenos en caso de anticoncepción hormonal.
En voluntarios sanos, el valproato desplazó al diazepam de sus sitios de unión con la albúmina plasmática e inhibió su metabolismo. En la terapia de combinación, la concentración de diazepam libre puede aumentar, mientras que el aclaramiento plasmático y el volumen de distribución de la fracción libre de diazepam pueden reducirse (por 25% y 20% respectivamente), sin embargo, la vida media permanece sin cambios.
En sujetos sanos, el tratamiento concomitante con valproato y lorazepam resultó en una reducción del aclaramiento plasmático de lorazepam en más del 40%.
Se ha producido ausencia en pacientes con antecedentes de epilepsia convulsiva de ausencia después de un tratamiento combinado de ácido valproico y clonazepam.
Después del tratamiento concomitante con ácido valproico, sertralina y risperidona, se desarrolló catatonia en un paciente con trastorno esquizoafectivo.
- Quetiapina
La administración concomitante de valproato y quetiapina puede aumentar el riesgo de neutropenia / leucopenia.
La ingesta concomitante de alimentos no afecta significativamente la biodisponibilidad del valproato de sodio cuando se administra como gránulos de Depakin de liberación modificada.
Advertencias Es importante saber que:
Niñas / Adolescentes / Mujeres en edad fértil / Embarazo:
Depakin no debe administrarse a niñas, adolescentes, mujeres en edad fértil y mujeres embarazadas, a menos que los tratamientos alternativos sean ineficaces o no se toleren, debido a su alto potencial teratogénico y al riesgo de trastornos del desarrollo en lactantes expuestos al útero al valproato. Los riesgos y beneficios deben reconsiderarse cuidadosamente durante las reevaluaciones regulares del tratamiento, en la pubertad y con carácter de urgencia cuando una mujer en edad fértil tratada con los planes Depakin o queda embarazada.
Las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces durante el tratamiento y ser informadas de los riesgos asociados con el uso de Depakin durante el embarazo (ver "Embarazo").
El médico que prescribe debe asegurarse de que el paciente reciba información completa sobre los riesgos, así como materiales relevantes, como un folleto de información para el paciente, para ayudarla a comprender los riesgos.
En particular, el prescriptor debe asegurarse de que el paciente comprenda:
- La naturaleza y el alcance de los riesgos de exposición durante el embarazo, en particular los riesgos teratogénicos y los riesgos relacionados con los trastornos del desarrollo.
- La necesidad de utilizar un método anticonceptivo eficaz.
- La necesidad de una revisión regular del tratamiento.
- La necesidad de consultar a su médico rápidamente si cree que se está quedando embarazada o existe la posibilidad de un embarazo.
En las mujeres que planean quedarse embarazadas, se debe hacer todo lo posible para cambiar a un tratamiento alternativo adecuado antes de la concepción, si es posible (ver "Embarazo").
La terapia con valproato solo debe continuarse después de una reevaluación de los beneficios y riesgos del tratamiento con valproato para el paciente por parte de un médico con experiencia en el tratamiento de la epilepsia o el trastorno bipolar.
Un pequeño número de pacientes en tratamiento con fármacos antiepilépticos como el valproato han desarrollado pensamientos de autolesión o suicidio. Si, en cualquier momento, tiene estos pensamientos, póngase en contacto con su médico inmediatamente.
No se recomienda el alcohol durante el tratamiento con valproato. Dado que el valproato se excreta principalmente a través de los riñones, en parte como cuerpos cetónicos, la prueba de excreción de cuerpos cetónicos puede dar resultados falsos positivos en pacientes diabéticos.
HEPATOPATÍAS
- Condiciones de aparición
Se ha informado de daño hepático excepcionalmente severo y en ocasiones ha sido fatal.
La experiencia en epilepsia ha indicado que los pacientes con mayor riesgo, especialmente en casos de terapia anticonvulsiva múltiple, son bebés y niños menores de 3 años con formas severas de epilepsia, particularmente aquellos con daño cerebral, retraso mental y (o) con trastornos metabólicos congénitos. o enfermedad degenerativa.
Si el Médico considera imprescindible administrar el fármaco a niños menores de tres años para el tratamiento de un tipo de epilepsia sensible al valproato, a pesar del riesgo de enfermedad hepática, el uso de Depakin debe realizarse solo para reducir este riesgo. A la edad de 3 años, la incidencia se reduce significativamente y disminuye progresivamente con la edad.
En la mayoría de los casos, se produjo daño hepático durante los primeros 6 meses de tratamiento.
- Sintomatología
Los síntomas clínicos son fundamentales para un diagnóstico precoz. En particular, se deben considerar dos tipos de manifestaciones que pueden preceder a la ictericia, especialmente en pacientes en riesgo (ver "Condiciones de inicio"):
- las convulsiones reaparecen en pacientes epilépticos
- síntomas inespecíficos, generalmente de inicio rápido, como astenia, anorexia, letargo, somnolencia, a veces asociados con vómitos repetidos y dolor abdominal.
Se debe advertir a los pacientes (o sus padres si son niños) que notifiquen a su médico de inmediato si se presenta alguno de los síntomas anteriores. Además de los controles clínicos, deben realizarse controles químicos sanguíneos inmediatos de la función hepática.
- Detección
Se debe controlar la función hepática antes del inicio de la terapia y periódicamente durante los primeros 6 meses. Entre los análisis habituales, los más relevantes son los que reflejan la síntesis de proteínas, especialmente el tiempo de protrombina. Confirmación de un porcentaje de actividad de protrombina. Particularmente bajo, especialmente si asociado con otros hallazgos biológicos anormales (disminución significativa de fibrinógeno y factores de coagulación; aumento de los niveles de bilirrubina y transaminasas SGOT, SGPT, gamma-GT, lipasa, alfa-amilasa, glucemia) requiere la interrupción del tratamiento con valproato. Como precaución en caso de que se tomen al mismo tiempo, también se deben suspender los salicilatos, ya que se metabolizan por la misma vía.
Cuatro semanas después del inicio del tratamiento, deben controlarse las pruebas de laboratorio para los parámetros de coagulación como INR y PTT, SGOT, SGPT, bilirrubina y amilasa.
En los niños sin síntomas clínicos anormales, se deben controlar los recuentos sanguíneos que incluyen trombocitos, SGOT y SGPT en cada visita.
PANCREATITAS
Muy raramente se han notificado casos de pancreatitis grave que puede ser mortal. Los niños más pequeños corren un riesgo especial. El riesgo disminuye con la edad. Los ataques graves, los trastornos neurológicos o la polifarmacia anticonvulsiva pueden ser factores de riesgo. La insuficiencia hepática concomitante con pancreatitis aumenta el riesgo de desenlace fatal. Los pacientes que experimentan dolor abdominal agudo deben ser atendidos inmediatamente por un médico. En caso de pancreatitis, se debe suspender el tratamiento con valproato.
FERTILIDAD, EMBARAZO Y LACTANCIA
Consulte a su médico o farmacéutico antes de tomar cualquier medicamento.
Depakin no debe administrarse a niñas, adolescentes, mujeres en edad fértil y mujeres embarazadas, a menos que otros tratamientos sean ineficaces o no se toleren. Las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces durante el tratamiento. En las mujeres que planean quedarse embarazadas, se debe hacer todo lo posible para cambiar a un tratamiento alternativo adecuado antes de la concepción, si es posible.
El embarazo
Riesgo de exposición durante el embarazo vinculado al valproato
Tanto el valproato solo como el valproato en politerapia se asocian con resultados anormales del embarazo. Los datos disponibles sugieren que la polifarmacia antiepiléptica, incluido el valproato, se asocia con un mayor riesgo de malformaciones congénitas en comparación con el valproato solo.
Malformaciones congénitas
Los datos de un metanálisis (que incluyó registros y estudios de cohortes) mostraron que el 10,73% de los hijos de mujeres epilépticas expuestas a valproato en monoterapia durante el embarazo padecen malformaciones congénitas (IC del 95%: 8,16 -13,29). Existe un mayor riesgo de malformaciones mayores que en la población general, para lo cual el riesgo es aproximadamente del 2-3%. El riesgo depende de la dosis, pero no se puede establecer una dosis umbral por debajo de la cual no existe riesgo.
Los datos disponibles demuestran una "mayor incidencia de malformaciones mayores y menores. Los tipos más comunes de malformaciones incluyen defectos del tubo neural, dismorfismo facial, labio leporino y paladar hendido, craneosinostosis, defectos cardíacos, renales y urogenitales, defectos de las extremidades (incluida la aplasia). Radio bilateral ) y múltiples anomalías que afectan a los distintos sistemas del organismo.
Trastornos del desarrollo
Los datos demostraron que la exposición al valproato en el útero puede tener efectos adversos en el desarrollo mental y físico de los niños expuestos. El riesgo parece depender de la dosis pero, según los datos disponibles, no se puede establecer una dosis umbral por debajo del umbral. No hay riesgo El período exacto de gestación con riesgo de estos efectos es incierto y no se puede excluir la posibilidad de riesgo durante el embarazo.
Los estudios de niños en edad preescolar expuestos en el útero al valproato muestran que hasta un 30-40% experimenta retrasos en el desarrollo temprano, como retraso en el habla y la marcha, disminución de la capacidad intelectual, deficiencias del lenguaje (habla y comprensión) y problemas de memoria.
El cociente intelectual (CI) medido en niños en edad escolar (6 años) con antecedentes de exposición al valproato en el útero fue en promedio 7-10 puntos más bajo que el de los niños expuestos a otros antiepilépticos. Aunque no se puede excluir el papel de los factores de confusión, existe evidencia en niños expuestos al valproato de que el riesgo de deterioro intelectual puede ser independiente del coeficiente intelectual materno.
Hay datos limitados sobre los resultados a largo plazo.
Los datos disponibles demuestran que los niños expuestos al valproato en el útero tienen un mayor riesgo de trastornos del espectro autista (aproximadamente tres veces) y autismo infantil (aproximadamente cinco veces) que la población general del estudio.
Los datos limitados sugieren que los niños expuestos al valproato en el útero pueden tener más probabilidades de desarrollar síntomas de trastorno por déficit de atención / hiperactividad (TDAH).
Niñas, adolescentes y mujeres en edad fértil (ver arriba y "Advertencias especiales")
Si una mujer desea planificar un embarazo
- Durante el embarazo, las convulsiones tónico-clónicas maternas y el estado epiléptico hipóxico pueden conllevar un riesgo particular de muerte para la madre y el feto.
- La terapia con valproato debe reevaluarse en mujeres que planean quedar embarazadas o embarazadas.
- En las mujeres que planean quedarse embarazadas, se debe hacer todo lo posible para cambiar a un tratamiento alternativo adecuado antes de la concepción, si es posible.
La terapia con valproato no debe suspenderse sin una reevaluación de los beneficios y riesgos del tratamiento con valproato para el paciente por parte de un médico con experiencia en el manejo de la epilepsia o el trastorno bipolar. Y beneficios, el tratamiento con valproato se continúa durante el embarazo, se recomienda:
- Use la dosis efectiva más baja y divida la dosis diaria de valproato en varias dosis pequeñas para tomarlas a lo largo del día. El uso de una formulación de liberación prolongada puede ser preferible al tratamiento con otras formulaciones para evitar concentraciones plasmáticas máximas altas. La dosis diaria debe administrarse en varias dosis pequeñas a lo largo del día en mujeres que pueden quedar embarazadas y ciertamente entre los días 20 y 40 después de la concepción. Además, las concentraciones plasmáticas deben controlarse regularmente, considerando la posibilidad de fluctuaciones considerables que pueden ocurrir durante el embarazo incluso con una dosis constante.
- La suplementación con ácido fólico antes del embarazo podría reducir el riesgo de defectos del tubo neural comunes a todos los embarazos. Sin embargo, la evidencia disponible no sugiere que prevenga los defectos de nacimiento o las malformaciones debidas a la exposición al valproato.
- Establecer un seguimiento prenatal especializado para detectar la posible aparición de defectos del tubo neural u otras malformaciones. Las mujeres en edad fértil deben ser informadas de los riesgos y beneficios de usar DEPAKIN durante el embarazo.
Riesgos para el recién nacido
- Muy raramente, se han notificado casos de síndrome hemorrágico en recién nacidos cuyas madres tomaron valproato durante el embarazo. Este síndrome hemorrágico está relacionado con trombocitopenia, hipofibrinogenemia y / o disminución de otros factores de coagulación. También se ha informado afibrinogenemia y podría ser fatal. Sin embargo, este síndrome debe distinguirse de la disminución de los factores de vitamina K. inducida por fenobarbital y por inducción de enzimas. En consecuencia, en los recién nacidos deben examinarse los recuentos de plaquetas, el nivel de fibrinógeno en plasma, las pruebas de coagulación y los factores de coagulación.
- Se han notificado casos de hipoglucemia en lactantes cuyas madres tomaron valproato en el tercer trimestre del embarazo.
- Ha habido informes de hipotiroidismo en recién nacidos cuyas madres tomaron valproato durante el embarazo.
- El síndrome de abstinencia (p. Ej., En particular, agitación, irritabilidad, hiperexcitabilidad, nerviosismo, hipercinesia, alteraciones de la tonicidad, temblores, convulsiones y trastornos de la alimentación) puede surgir en recién nacidos cuyas madres han tomado valproato en el último trimestre del embarazo.
El tratamiento con ácido valproico durante el embarazo no debe interrumpirse sin consultar a su médico, así como cualquier interrupción abrupta del tratamiento o reducción incontrolada de la dosis. Esto podría provocar convulsiones en la mujer embarazada, lo que podría dañar a la madre y / o al feto.
El embarazo
El valproato se excreta en la leche materna a una concentración que oscila entre el 1% y el 10% de los niveles séricos maternos. Se han observado alteraciones hematológicas en lactantes de mujeres tratadas (ver "Reacciones adversas").
Se debe decidir si interrumpir la lactancia o interrumpir / abstenerse del tratamiento con Depakin teniendo en cuenta el beneficio de la lactancia para el niño y el beneficio del tratamiento para la mujer.
Fertilidad
Se han informado casos de amenorrea, ovario poliquístico y aumento de los niveles de testosterona en mujeres que usan valproato (consulte "Efectos secundarios"). La administración de valproato también puede afectar la fertilidad en los hombres (ver "Reacciones adversas"). Los casos clínicos indican que las disfunciones de la fertilidad son reversibles después de la interrupción del tratamiento.
Efectos sobre la capacidad para conducir y utilizar máquinas.
En caso de administración concomitante con barbitúricos u otros fármacos con actividad depresora del sistema nervioso central, en algunos sujetos se pueden encontrar manifestaciones de astenia, somnolencia o confusión, que pueden alterar así la respuesta a la capacidad para conducir un vehículo, utilizar maquinaria o realizar actividades. relacionada con el riesgo de caída o accidente, la capacidad se ve afectada independientemente de la enfermedad subyacente.
Las mismas manifestaciones se pueden observar después de beber bebidas alcohólicas. Se debe advertir de ello a aquellos sujetos que, durante el trámite, pudieran conducir vehículos o atender operaciones que requieran integridad del grado de supervisión.
Posología y forma de empleo Cómo usar Depakin: Posología
Entre las formas farmacéuticas orales, las más adecuadas para su administración en niños menores de 11 años son la solución oral y el granulado.
DEPAKIN Modified Release Granules es una forma farmacéutica adecuada para todos, especialmente niños (si pueden tragar alimentos blandos), adultos con dificultades para tragar y ancianos.
En función de la cantidad de principio activo, los sobres de 50 mg y 100 mg están reservados para los niños.
DEPAKIN Modified Release Granules es una formulación de liberación controlada de Depakin que reduce las concentraciones máximas y asegura concentraciones plasmáticas más regulares a lo largo del día.
Tratamiento de la epilepsia
La dosificación diaria debe basarse en la edad y el peso corporal; sin embargo, también debe tenerse en cuenta la amplia sensibilidad individual al valproato.
No se ha establecido una correlación definitiva entre la dosis diaria, la concentración sérica y el efecto terapéutico, y la dosis óptima debe determinarse esencialmente de acuerdo con la respuesta clínica; se puede considerar la determinación de los niveles plasmáticos de ácido valproico además de la monitorización clínica. de ataques no se logra o cuando se sospechan eventos adversos. El rango terapéutico generalmente está entre 40-100 mg / L (300-700 µmol / L).
La dosis establecida debe dividirse en 2 administraciones diarias.
Inicio de la terapia con gránulos de DEPAKIN de liberación modificada (administración oral)
- En pacientes no tratados con otros fármacos antiepilépticos, la dosis debe aumentarse preferiblemente por niveles de dosis sucesivos, a intervalos de 2-3 días, para alcanzar el óptimo en aproximadamente una semana.
- En pacientes que ya están en tratamiento con fármacos antiepilépticos, la sustitución por DEPAKIN granulado de liberación modificada debe ser gradual, alcanzando la dosis óptima en unas dos semanas, reduciendo y luego interrumpiendo los demás tratamientos.
- La adición de otro fármaco antiepiléptico debe hacerse gradualmente, según sea necesario (consulte "Interacciones").
Administración oral de DEPAKIN granulado de liberación modificada: consideraciones prácticas
Dosis
La dosis diaria inicial suele ser de 10 a 15 mg / kg, luego las dosis se ajustan a la dosis óptima (consulte "Inicio de la terapia con gránulos de DEPAKIN de liberación modificada").
Por lo general, se encuentra entre 20 y 30 mg / kg. No obstante, si no se consigue el control de los ataques con esta posología, es posible incrementar aún más la dosis, de forma adecuada; Los pacientes deben ser monitoreados de cerca cuando reciban dosis diarias superiores a 50 mg / kg (ver "Precauciones de uso").
En ninos la dosis de mantenimiento habitual es de unos 30 mg / kg por día.
En adultos la dosis de mantenimiento habitual está entre 20-30 mg / kg por día.
En la vejezAunque la farmacocinética de los gránulos de liberación modificada DEPAKIN está modificada, la importancia clínica es limitada y la dosis debe determinarse en función del control de las convulsiones.
En pacientes con insuficiencia renal o hipoproteinemia, se debe considerar el aumento de ácido valproico libre en suero y, si es necesario, se debe reducir la dosis.
Episodios de manía relacionados con el trastorno bipolar.
En adultos:
La dosis diaria debe ser establecida y controlada individualmente por el médico.
La dosis diaria inicial recomendada es de 750 mg. Además, en los ensayos clínicos, una dosis inicial de 20 mg de valproato / kg de peso corporal también mostró un perfil de seguridad aceptable. Las formulaciones de liberación prolongada pueden administrarse una o dos veces al día. La dosis debe aumentarse lo más rápidamente posible para alcanzar la dosis más baja. dosis terapéutica con la que se consigue el efecto clínico deseado. La dosis diaria debe adaptarse a la respuesta clínica para establecer la dosis efectiva más baja para el paciente individual. La dosis diaria promedio generalmente varía entre 1000 y 2000 mg de valproato. Los pacientes que reciben una dosis diaria superior a 45 mg / kg de peso corporal deben ser controlados de cerca.
La continuación del tratamiento en los episodios maníacos relacionados con el trastorno bipolar debe establecerse de forma individual a la dosis eficaz más baja.
Niños y adolescentes:
Depakin no debe utilizarse en niños y adolescentes menores de 18 años para el tratamiento de la manía.
Niñas, adolescentes, mujeres en edad fértil y mujeres embarazadas
Depakin debe ser iniciado y supervisado por un especialista con experiencia en el tratamiento de la epilepsia o el trastorno bipolar. El tratamiento solo debe iniciarse si otros tratamientos son ineficaces o no se toleran (ver "Advertencias especiales - Embarazo") y los beneficios y riesgos deben reconsiderarse cuidadosamente. Durante las reevaluaciones regulares del tratamiento. Preferiblemente, Depakin debe prescribirse como monoterapia y en la dosis efectiva más baja, si es posible como una formulación de liberación prolongada para evitar concentraciones plasmáticas máximas altas. La dosis diaria debe dividirse en al menos dos dosis únicas.
Método de administración para ambas indicaciones
Los gránulos de liberación modificada DEPAKIN se presentan en gránulos esféricos insípidos y deben administrarse preferiblemente distribuidos en alimentos blandos (yogur, fruta cocida, quesos frescos, etc.) o bebidas (jugo de naranja, etc.) fríos oa temperatura ambiente.
Los gránulos de liberación modificada DEPAKIN no deben administrarse con alimentos o bebidas tibios o calientes (sopas, café, té, etc.).
Los gránulos de liberación modificada DEPAKIN no deben introducirse en el biberón, ya que pueden bloquear la tetina.
Cuando se toma con líquidos, se recomienda enjuagar el vaso con una pequeña cantidad de agua porque algunos gránulos pueden pegarse al vaso.
Si lo prefiere, los gránulos pueden colocarse directamente en la boca y tragarse con agua o bebidas frías oa temperatura ambiente.
La preparación debe tragarse inmediatamente y no debe masticarse. No debe almacenarse para su uso posterior.
Teniendo en cuenta el proceso de liberación y la naturaleza de los excipientes de la formulación, la matriz inerte de los gránulos no es absorbida por el tracto digestivo y se elimina con las heces después de la liberación del ingrediente activo.
Sobredosis Qué hacer si ha tomado demasiado Depakin
En caso de ingestión / ingesta de una dosis excesiva de Depakin, notifique a su médico inmediatamente o acuda al hospital más cercano.
Signos y síntomas
A niveles séricos terapéuticos (50-100 µg / ml), el ácido valproico tiene una toxicidad relativamente baja. Muy raramente, se ha producido intoxicación aguda por ácido valproico a niveles séricos superiores a 100 µg / ml en adultos y niños.
Los signos de sobredosis aguda masiva generalmente incluyen coma con hipotonía muscular, hiporreflexia, miosis, función respiratoria alterada, acidosis metabólica, hipotensión, trastornos cardiovasculares, colapso / shock circulatorio e hipernatremia. La presencia de sodio en la formulación de valproato puede provocar hipernatremia cuando se ingiere en sobredosis.
Tanto en adultos como en niños, los niveles séricos elevados provocan trastornos neurológicos anormales, como una mayor tendencia a sufrir convulsiones y cambios de comportamiento.
Se han producido muertes después de una sobredosis masiva, sin embargo, el pronóstico de intoxicación es generalmente favorable. Sin embargo, los síntomas pueden ser variables y se han informado convulsiones en presencia de niveles plasmáticos muy altos.
Se han notificado casos de hipertensión intracraneal relacionada con edema cerebral.
Tratamiento
No se conoce ningún antídoto específico. Por tanto, el tratamiento clínico de la sobredosis debe limitarse a medidas generales destinadas a eliminar toxinas y apoyar las funciones vitales.
Las medidas a tomar a nivel hospitalario deben ser sintomáticas: lavado gástrico, que puede ser útil hasta 10-12 horas después de la ingestión; monitorización cardíaca y respiratoria. La naloxona se ha utilizado con éxito en algunos casos aislados. Sobredosis, hemodiálisis y hemoperfusión se han utilizado con éxito.
En caso de ingestión / ingesta accidental de una sobredosis de DEPAKIN, notifique a su médico inmediatamente o acuda al hospital más cercano.
SI TIENE ALGUNA DUDA SOBRE EL USO DE DEPAKIN, CONSULTE A SU MÉDICO O FARMACÉUTICO
Efectos secundarios ¿Cuáles son los efectos secundarios de Depakin?
Como todos los medicamentos, DEPAKIN puede producir efectos adversos, aunque no todas las personas los sufran.
Muy frecuentes: ≥ 1/10
Frecuentes: ≥ 1/100,
Poco frecuentes: ≥ 1/1000,
Raras: ≥ 1/10000,
Muy raro:
- Trastornos congénitos, familiares y genéticos
Malformaciones congénitas y trastornos del desarrollo (consulte "Advertencias especiales - Embarazo").
- Trastornos hepatobiliares
Frecuentes: puede producirse una disfunción hepática grave (a veces mortal), es independiente de la dosis. En los niños, particularmente en terapia combinada con otros antiepilépticos, el riesgo de daño hepático aumenta significativamente (ver "Advertencias especiales").
- Desórdenes gastrointestinales
Muy frecuentes: náuseas.
Frecuentes: vómitos, enfermedad de las encías (principalmente hiperplasia gingival), estomatitis, dolor abdominal superior, diarrea ocurren con frecuencia en algunos pacientes al inicio del tratamiento, pero generalmente desaparecen después de unos días sin interrumpir el tratamiento.
Poco frecuentes: sialorrea, pancreatitis, a veces mortal (ver "Advertencias especiales" y Precauciones de uso).
- Patologías endocrinas
Poco frecuentes: síndrome de secreción inadecuada de ADH (SIADH), hiperandrogenismo (hirsutismo, virilismo, acné, alopecia masculina y / o aumento de las hormonas andrógenas).
Raras: hipotiroidismo (ver "Advertencias especiales").
- Trastornos del metabolismo y de la nutrición.
Frecuentes: hiponatremia, aumento o pérdida de peso dependiente de la dosis, aumento del apetito y pérdida del apetito. En un estudio clínico con 75 niños, se observó una reducción de la actividad de la biotinidasa durante el tratamiento con medicamentos que contienen ácido valproico. También hubo informes de deficiencia de biotina.
Raras: hiperamonemia.
La hiperamonemia aislada moderada puede ocurrir sin pruebas de función hepática anormales y no debe ser una causa para la interrupción del tratamiento. Sin embargo, en el curso de la monoterapia o politerapia (fenobarbital, carbamazepina, fenitoína, topiramato) puede haber un síndrome agudo de encefalopatía hiperamonémica, con función hepática normal y ausencia de citólisis. El síndrome de encefalopatía hiperamonémica inducida por valproato se presenta en forma aguda y se caracteriza por pérdida de conciencia, estupor, debilidad muscular (hipotensión muscular), alteraciones motoras (discinesia coreoide), cambios generalizados graves en el EEG y signos neurológicos focales y generales con mayor frecuencia. de convulsiones. Puede aparecer unos días o semanas después del inicio de la terapia y regresa con la suspensión del valproato.La encefalopatía no está relacionada con la dosis y los cambios en el EEG se caracterizan por la aparición de ondas lentas y aumento de las descargas epilépticas.
- Neoplasias benignas, malignas y no especificadas (incluidos quistes y pólipos)
Raras: síndrome mielodisplásico.
- Trastornos del sistema nervioso
Muy frecuentes: temblor.
Frecuentes: parestesia dosis dependiente, trastornos extrapiramidales (incapacidad para permanecer sentado, rigidez, temblores, movimientos lentos, movimientos involuntarios, contracciones musculares). estupor, temblor postural, somnolencia, convulsiones, memoria insuficiente, dolor de cabeza, nistagmo, mareos a los pocos minutos de la administración intravenosa, que desaparecen espontáneamente a los pocos minutos.
Poco frecuentes: espasticidad, ataxia, especialmente al inicio del tratamiento, coma, encefalopatía, letargo, parkinsonismo reversible.
Raras: demencia reversible asociada con atrofia cerebral reversible, alteraciones cognitivas, estados de confusión. El estupor y el letargo, que a veces conducen a un coma transitorio (encefalopatía), fueron casos aislados o se asociaron con una mayor incidencia de convulsiones durante el tratamiento y retrocedieron con la interrupción del tratamiento o la reducción de la dosis. Estos casos se han notificado principalmente durante la terapia de combinación (especialmente con fenobarbital o topiramato) o después de un aumento brusco de las dosis de valproato.
Se ha informado de sedación.
- Desórdenes psiquiátricos
Frecuentes: estado de confusión, alucinaciones, agresión *, agitación *, alteración de la atención *.
Poco frecuentes: irritabilidad, hiperactividad y confusión, especialmente al inicio del tratamiento (ocasionalmente agresión, alteraciones del comportamiento).
Raras: comportamiento anormal *, hiperactividad psicomotora *, trastornos del aprendizaje *
* Estos efectos secundarios se han observado principalmente en niños.
- Trastornos del sistema sanguíneo y linfático.
Frecuentes: anemia, trombocitopenia
Poco frecuentes: neutropenia, leucopenia o pancitopenia, hipoplasia de glóbulos rojos. Edema periférico, sangrado
Raras: insuficiencia de la médula ósea, incluida la aplasia pura de la médula ósea que afecta a los glóbulos rojos.
Agranulocitosis, anemia macrocítica, macrocitosis.
Pruebas de diagnóstico
Frecuentes: aumento de peso. Dado que el aumento de peso es un factor de riesgo para el síndrome de ovario poliquístico, debe controlarse cuidadosamente (consulte "Precauciones de uso").
Raras: factores de coagulación disminuidos (al menos uno), deficiencia del factor VIII (factor de von Willebrand), pruebas de coagulación anormales (como prolongación del tiempo de protrombina, prolongación del tiempo de tromboplastina parcial activada, prolongación del tiempo de trombina, INR prolongado) (ver también " El embarazo").
Ha habido informes aislados de disminución del fibrinógeno.
Deficiencia de biotina / biotinidasa.
- Trastornos de la piel y del tejido subcutáneo
Frecuentes: hipersensibilidad, alopecia transitoria y (o) relacionada con la dosis.
Poco frecuentes: angioedema, erupción cutánea, cambios en el cabello (como estructura anormal del cabello, cambios en el color del cabello, crecimiento anormal del cabello).
Raras: necrólisis epidérmica tóxica, síndrome de Stevens-Johnson, eritema multiforme. Síndrome de fiebre de drogas con eosinofilia y síntomas sistémicos (DRESS), reacciones alérgicas.
- Enfermedades del aparato reproductor y la mama.
Niveles elevados de testosterona. Ha habido informes de frecuencia de ovario poliquístico en pacientes que han tenido un aumento de peso significativo.
Frecuentes: dismenorrea,
Poco frecuentes: amenorrea.
Raras: infertilidad masculina.
- Patologías vasculares
Frecuentes: hemorragia (ver "Precauciones de uso" y "Advertencias especiales")
Poco frecuentes: vasculitis.
- Desordenes generales y condiciones administrativas del sitio
Poco frecuentes: hipotermia.
- Trastornos del oído y del laberinto.
Frecuentes: sordera, acúfenos.
- Trastornos respiratorios, torácicos y mediastínicos
Poco frecuentes: derrame pleural.
- Trastornos renales y urinarios.
Poco frecuentes: insuficiencia renal.
Raras: enuresis, nefritis tubulointersticial, síndrome de Fanconi reversible, el mecanismo de acción aún no está claro.
- Trastornos del sistema inmunológico.
Raras: lupus eritematoso sistémico, rabdomiólisis (ver Precauciones de uso).
- Trastornos musculoesqueléticos y del tejido conjuntivo
Se han notificado casos de disminución de la densidad mineral ósea, osteopenia, osteoporosis y fracturas en pacientes en tratamiento a largo plazo con Depakin. El mecanismo por el cual Depakin afecta el metabolismo óseo sigue sin estar claro.
En cuanto a los efectos indeseables relacionados con el S.N.C. y el posible riesgo teratogénico, estos podrían tener una "menor incidencia que los que se presentan tras la administración de Depakin. De hecho, DEPAKIN granulado de liberación modificada tiene un perfil plasmático más regular, con menores fluctuaciones en las concentraciones de ácido valproico debido a la reducción de niveles en sangre, picos (Cmax) y con niveles de "cable" sin cambios.
El cumplimiento de las instrucciones contenidas en el prospecto reduce el riesgo de reacciones adversas.
Notificación de efectos secundarios
Si experimenta efectos adversos, consulte a su médico o farmacéutico, incluidos los posibles efectos adversos que no aparecen en este prospecto. Las reacciones adversas también pueden notificarse directamente a través del sistema nacional de notificación en "https://www.aifa.gov.it/content/segnalazioni-reazioni-avverse." Información sobre la seguridad de este medicamento.
Caducidad y retención
Caducidad: consulte la fecha de caducidad impresa en el paquete.
La fecha de caducidad se refiere al producto en envases intactos y correctamente almacenados.
Advertencia: no use el medicamento después de la fecha de caducidad que se muestra en el paquete
No conservar por encima de 25 ° C.
Almacenar en el paquete original, proteger el medicamento de la humedad o fuentes de calor.
No refrigerar o congelar
Mantenga este medicamento fuera del alcance y la vista de los niños.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que no necesita, ya que esto ayudará a proteger el medio ambiente.
Prospecto fuente: AIFA (Agencia Italiana de Medicamentos). Contenido publicado en enero de 2016. Es posible que la información presente no esté actualizada.
Para tener acceso a la versión más actualizada, es recomendable acceder al sitio web de la AIFA (Agencia Italiana de Medicamentos). Descargo de responsabilidad e información útil.
01.0 NOMBRE DEL MEDICAMENTO
CLENIL - POLVO PARA INHALACIÓN
02.0 COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Clenil 100 mcg polvo para inhalación
Cada entrega contiene:
Principio activo: dipropionato de beclometasona 100 mcg.
Clenil 200 mcg polvo para inhalación
Cada entrega contiene:
Principio activo: dipropionato de beclometasona 200 mcg.
Clenil 400 mcg polvo para inhalación
Cada entrega contiene:
Principio activo: dipropionato de beclometasona 400 mcg.
Para consultar la lista completa de excipientes, ver sección 6.1.
03.0 FORMA FARMACÉUTICA
Polvo de inhalación en inhalador Pulvinal.
04.0 INFORMACIÓN CLÍNICA
04.1 Indicaciones terapéuticas
Control de la evolución de enfermedades asmáticas y broncostenosis.
04.2 Posología y forma de administración
Adultos
Una inhalación de Clenil 400mcg en polvo dos veces al día o una inhalación de Clenil 200mcg en polvo 3-4 veces al día.
En pacientes que requieran dosis más altas para controlar la enfermedad del asma, la dosis puede aumentarse a dos inhalaciones dos veces al día de Clenil 400 mcg en polvo.
Niños
Una inhalación de Clenil 100mcg en polvo 2-4 veces al día o una inhalación de Clenil 200mcg en polvo dos veces al día.
Para obtener buenos resultados, la preparación debe utilizarse con regularidad, incluso durante las fases asintomáticas.
No se requieren ajustes de dosis en pacientes de edad avanzada o pacientes con insuficiencia hepática o renal.
El polvo para inhalación de Clenil es solo para uso por inhalación.
Instrucciones de uso
Lea atentamente las siguientes instrucciones para un uso correcto. Si es necesario, comuníquese con su médico para obtener explicaciones más detalladas.
Clenil es un polvo para inhalación a base de dipropionato de beclometasona micronizado mezclado con un "portador", contenido en un dispositivo inhalador multidosis. El sistema de suministro no requiere propulsores y no requiere coordinación entre el suministro y la inhalación.
El inhalador de polvo debe almacenarse en un lugar seco a temperatura ambiente.
No retire la tapa protectora hasta el momento de su uso.
PARA) Apertura
1) Desatornille la tapa protectora. Antes de usar, verifique que la boquilla esté limpia. Si es necesario, límpiela con una toalla de papel sin pelusa o un paño suave. Antes de girar el inhalador, sosténgalo en posición vertical y golpéelo suavemente sobre una superficie dura para nivelar el polvo en la Cámara.
B) Cargando
2) Sosteniendo el inhalador en posición vertical, presione el botón marrón de la boquilla con una mano y con la otra gire el cuerpo del inhalador en sentido antihorario (media vuelta) tanto como sea posible, con el orificio en la boquilla colocado exactamente en el punto rojo (posición de carga de la dosis).
3) Mientras sostiene el inhalador en posición vertical, gire el cuerpo del inhalador en el sentido de las agujas del reloj (media vuelta) hasta que escuche un "clic", con el orificio colocado exactamente en el punto verde (posición de administración de la dosis).
C) Administración
4) Exhale profundamente con calma, no a través del inhalador.
5) Coloque la boquilla entre los labios, sosteniendo el inhalador en posición vertical, e inhale por la boca lo más rápido y profundamente posible, contenga la respiración durante unos segundos.
D) Cierre
6) Retire el inhalador de la boca y vuelva a enroscar la tapa protectora.
Consejos generales
Mantenga siempre el inhalador en posición vertical desde la fase de carga de la dosis hasta la inhalación.
Si se va a tomar una dosis correspondiente a 2 inhalaciones, es necesario rotar el inhalador cada vez como se describe arriba en el punto B antes de inhalar.
Durante el uso, el nivel del polvo disminuirá gradualmente en el cuerpo transparente del dispositivo. Cuando los rayos rojos en la parte inferior del inhalador se hagan visibles, debido al bajo nivel del polvo, el inhalador debe ser reemplazado ya que, a partir de ese En ese momento, no corregirá la entrega de la dosis, está más garantizada.
Después de inhalar la dosis y antes de cerrar el inhalador, verifique que el orificio de la boquilla esté colocado en el punto verde del cuerpo del inhalador.
La presencia de polvo en la boca tras la inhalación y una ligera sensación de sabor dulce son una confirmación de que la dosis se ha administrado correctamente y el principio activo ha llegado a los pulmones.
El inhalador contiene una cápsula desecante que asegura un correcto nivel de humedad dentro de la cámara de dispensación, el tapón siempre debe volver a colocarse después de usar el inhalador, además, el inhalador nunca debe colocarse cerca de fuentes de calor o humedad. Sin embargo, si el inhalador se almacena sin tapar durante períodos cortos (por ejemplo, 24 horas) a temperatura ambiente, la eficacia del producto no se ve afectada.
Si, en lugar de inhalar, sopla en el inhalador, esto no causa ningún problema: si lo hace, simplemente dé la vuelta al inhalador y vacíe el polvo de la cámara de dosificación. Debe proceder de la misma manera si cree que ha cargado dos o más dosis en la cámara.
Instrucciones de limpieza
Durante el uso, limpie regularmente la boquilla con un pañuelo de papel sin pelusa o un paño suave.
04.3 Contraindicaciones
Hipersensibilidad a las cortisonas oa alguno de los excipientes.
04.4 Advertencias especiales y precauciones de uso apropiadas
Se debe instruir a los pacientes sobre el uso correcto del inhalador y su método controlado para asegurarse de que el fármaco llegue a las áreas objetivo dentro de los pulmones. También se debe advertir a los pacientes que Clenil Polvo para inhalación debe tomarse regularmente en las dosis prescritas cada día durante períodos prolongados, incluso cuando los pacientes estén asintomáticos.
El polvo para inhalación de Clenil no es eficaz en los ataques de asma en curso en los que se requiere un broncodilatador inhalado de acción rápida. Se debe advertir a los pacientes que tengan este tipo de medicamento disponible.
El aumento en el uso de broncodilatadores, particularmente broncodilatadores inhalados de agonistas beta2 de acción corta, indica un empeoramiento del control de la enfermedad del asma. Si el paciente cree que el tratamiento sintomático con broncodilatadores de acción corta, la acción se vuelve menos efectiva o si usa más inhalaciones de lo habitual , se requiere un examen médico.
En esta situación, se debe volver a evaluar a los pacientes y se debe considerar la necesidad o la posibilidad de aumentar el tratamiento antiinflamatorio (por ejemplo, aumentar la dosis de corticosteroides inhalados o iniciar un ciclo con corticosteroides orales). Las exacerbaciones graves del asma deben tratarse de la forma convencional.
El tratamiento con Clenil polvo para inhalación no debe interrumpirse bruscamente.
Rara vez ocurre una supresión significativa de la función suprarrenal hasta dosis de 1500 mcg / día de dipropionato de beclometasona inhalado. Algunos pacientes tratados con 2000 mcg / día experimentaron reducciones en los niveles plasmáticos de cortisol. En tales pacientes, el riesgo de desarrollar supresión suprarrenal debe sopesarse con los beneficios terapéuticos, y deben tomarse precauciones para proporcionar cobertura de esteroides sistémicos en situaciones estresantes prolongadas (p. Ej., Cirugía electiva). La supresión prolongada del eje hipotalámico-pituitario-suprarrenal puede tener efectos sistémicos con los corticosteroides inhalados, especialmente cuando se prescriben en dosis altas durante períodos prolongados. Estos efectos son menos probables que con los corticosteroides orales. Posibles efectos Los trastornos sistémicos incluyen síndrome de Cushing, características cushingoides , supresión suprarrenal, retraso del crecimiento en niños y adolescentes, disminución de la densidad mineral ósea, cataratas, glaucoma y, más raramente, una serie de efectos psicológicos o conductuales que incluyen hiperactividad psicomotora, trastornos del sueño, ansiedad, depresión o agresión (especialmente en niños). Por tanto, es importante que la dosis de corticosteroides inhalados sea la dosis más baja posible con la que se mantenga un control eficaz del asma.
Se recomienda controlar periódicamente la altura de los niños tratados con corticosteroides inhalados. En el caso de crecimiento lento, se debe revisar la terapia para reducir, si es posible, la dosis del corticoesteroide inhalado hasta alcanzar la dosis mínima eficaz para mantener la dosis. control del asma. Además, se recomienda considerar la posibilidad de derivar al paciente a un pediatra especializado en enfermedades respiratorias.
Se debe prestar especial atención al traslado de pacientes de la terapia con esteroides sistémica continua, a largo plazo o en dosis altas a la terapia con dipropionato de beclometasona, ya que la recuperación de la función suprarrenal suprimida puede llevar un período de tiempo considerable. Clenil Powder para inhalación debe administrarse inicialmente mientras se continúa el tratamiento sistémico; después de aproximadamente una semana, cuando el paciente está estabilizado, los esteroides sistémicos pueden reducirse progresivamente. La magnitud de la reducción debe corresponder a la dosis de mantenimiento del esteroide sistémico Durante esta disminución del esteroide, la función suprarrenal debe controlarse regularmente.
Algunos pacientes experimentan malestar general durante la interrupción del tratamiento a pesar de que su función respiratoria permanece inalterada o incluso mejor. A menos que existan signos clínicos objetivos de insuficiencia suprarrenal, se debe alentar a estos pacientes a que continúen tomando Clenil en polvo inhalado y continúen descontinuando el esteroide sistémico.
Estas precauciones no deben aplicarse a pacientes en tratamiento con esteroides orales durante menos de 2 semanas. Un esteroide oral y un polvo Clenil inhalado pueden necesitar iniciarse simultáneamente en un paciente con síntomas de asma. Una vez que se ha logrado un buen control del asma (mediante la monitorización del flujo espiratorio máximo), se puede interrumpir el esteroide oral. De forma abrupta, de nuevo si se ha administrado por menos de 2 semanas. El tratamiento con Clenil Powder para inhalación debe continuarse para mantener el control de la enfermedad asmática.
Los pacientes que han dejado de tomar corticosteroides orales y tienen disfunción suprarrenal pueden necesitar un tratamiento adicional con esteroides sistémicos durante momentos de estrés, por ejemplo, en el caso de un empeoramiento del "ataque de asma", en el caso de infecciones del tórax, enfermedades graves concomitantes, cirugía, trauma, etc.
Reemplazar el tratamiento con esteroides sistémicos con terapia de inhalación puede resultar en alergias (como rinitis alérgica o eccema) previamente controladas por tratamiento sistémico. Estas alergias deben tratarse sintomáticamente con antihistamínicos y / o preparaciones locales, incluidos esteroides locales.
Al igual que con todos los corticosteroides inhalados, se debe tener especial cuidado en pacientes con tuberculosis pulmonar activa o inactiva, infecciones víricas, bacterianas y micóticas de los ojos, la boca y el tracto respiratorio. Puede ser necesaria una infección bacteriana del tracto respiratorio. Suspensión del tratamiento y terapia específica con antibióticos.
Este medicamento contiene aproximadamente 25 mg de lactosa monohidrato por dosis. Los pacientes con intolerancia hereditaria a galactosa, deficiencia de lactasa de Lapp o problemas de absorción de glucosa o galactosa no deben tomar este medicamento.
Mantenga este medicamento fuera del alcance y la vista de los niños.
04.5 Interacciones con otros medicamentos y otras formas de interacción
Debido a la concentración plasmática muy baja que se alcanza tras la administración inhalada, es poco probable que haya interacciones clínicamente significativas con otros fármacos. Sin embargo, podría producirse un aumento potencial de la exposición sistémica a la beclometasona cuando se administran concomitantemente inhibidores potentes de la enzima CYP3A4 (p. Ej., Ketoconazol, itraconazol, nelfinavir, ritonavir).
04.6 Embarazo y lactancia
No se ha establecido la seguridad del uso de dipropionato de beclometasona en el embarazo humano Los estudios de toxicología reproductiva en animales han revelado una mayor incidencia de daño fetal, cuya importancia se considera incierta en humanos. Dado que existe la posibilidad de supresión de la función adrenocortical en los recién nacidos después de un tratamiento prolongado, el beneficio para la madre debe sopesarse cuidadosamente con el riesgo para el feto.
Es razonable suponer que el medicamento está presente en la leche materna, pero a las dosis de inhalación utilizadas, la posibilidad de encontrar concentraciones significativas en la leche materna es baja.
Los bebés nacidos de madres que recibieron dosis sustanciales de corticosteroides inhalados durante el embarazo deben ser cuidadosamente observados para detectar hipoadrenalismo.
04.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han notificado efectos sobre la capacidad para conducir y utilizar máquinas.
04.8 Efectos indeseables
No se han notificado efectos indeseables graves tras la administración de Clenil Polvo para inhalación a la dosis recomendada.
Los eventos adversos se enumeran a continuación por sistema de clasificación de órganos y frecuencia. Las frecuencias se definen de la siguiente manera: muy frecuentes (≥1 / 10), frecuentes (≥1 / 100 a
Al igual que con otras terapias de inhalación, puede producirse un broncoespasmo paradójico con aumento inmediato de las sibilancias después de la administración. Esto debe tratarse inmediatamente con un broncodilatador inhalado de acción rápida. El tratamiento con Clenil Powder para inhalación debe suspenderse inmediatamente, evaluar al paciente y, si es necesario, instituirse una terapia alternativa.
Algunos pacientes experimentan candidiasis de la boca y la garganta (cándida) particularmente en dosis más altas.
Se recomienda enjuagar la boca con agua inmediatamente después de la inhalación. La candidiasis sintomática se puede tratar con terapia antifúngica tópica.
Los corticosteroides inhalados pueden tener efectos sistémicos, particularmente a dosis altas prescritas por períodos prolongados. Estos incluyen supresión de la corteza suprarrenal, retraso del crecimiento en niños y adolescentes, disminución de la densidad mineral ósea que conduce a osteoporosis, cataratas y glaucoma y hematomas simples en la piel, infecciones del tracto respiratorio inferior, incluida la neumonía, en pacientes de edad avanzada y enfermedad pulmonar obstructiva crónica (EPOC). .
04.9 Sobredosis
En caso de sobredosis no son necesarias intervenciones urgentes, la restauración de la función suprarrenal se consigue en pocos días y se puede verificar mediante la determinación de la cortisolemia.
El tratamiento con Clenil polvo para inhalación debe continuarse a las dosis recomendadas para el control del asma.
05.0 PROPIEDADES FARMACOLÓGICAS
05.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: glucocorticoide antiasmático para inhalación, código ATC: R03BA01.
Clenil Powder para inhalación contiene dipropionato de beclometasona como ingrediente activo, un corticosteroide con una fuerte actividad tópica antiinflamatoria y antialérgica en la mucosa del tracto respiratorio. En particular, el dipropionato de beclometasona ejerce una marcada acción antirreactiva a nivel bronquial, reduciendo el edema y la hipersecreción e inhibiendo la aparición del broncoespasmo. El dipropionato de beclometasona administrado por inhalación actúa exclusivamente sobre las estructuras del sistema respiratorio y está libre de las dosis recomendadas, de efectos sistémicos y acción inhibidora sobre la función cortico-adrenal.
05.2 Propiedades farmacocinéticas
Después de la inhalación de dipropionato de beclometasona, la fracción absorbida directamente en los pulmones es rápidamente metabolizada por el hígado a beclometasona-17-monopropionato y posteriormente al metabolito inactivo beclometasona alcohol.
05.3 Datos preclínicos sobre seguridad
Toxicidad aguda
DL50 (rata, por inhalación)> 2,68 mg / kg; (ratón, vía inhalada)> 4,93 mg / kg; (ratón, os)> 3000 mg / kg; (rata, os)> 1000 mg / kg.
Toxicidad por dosis repetidas (rata, nasal, 4 semanas)
Sin signos de toxicidad hasta una dosis de 220 mcg / kg / día.
La administración prolongada (1 año) por inhalación, a dosis muy superiores a las previstas en terapia, no provoca en el animal signos de sufrimiento en el tracto respiratorio.
06.0 INFORMACIÓN FARMACÉUTICA
06.1 Excipientes
Lactosa monohidrato, estearato de magnesio.
06.2 Incompatibilidad
No conocida.
06.3 Período de validez
3 años.
Este período está destinado a la especialidad debidamente almacenada y con el embalaje intacto.
06.4 Precauciones especiales de conservación
Mantenga siempre el inhalador bien cerrado con la tapa protectora.
No coloque el inhalador cerca de fuentes de calor o humedad.
06.5 Naturaleza del envase primario y contenido del envase.
Embalaje interno
Dispositivo inhalador multidosis pulmonar compuesto por boquilla, cuerpo transparente, base con desecante y capuchón protector.
Embalaje externo
Estuche de cartón estampado.
Clenil 100 mcg polvo para inhalación: inhalador de 100 inhalaciones
Clenil 200 mcg polvo para inhalación: inhalador de 100 inhalaciones
Clenil 400 mcg polvo para inhalación: inhalador de 100 inhalaciones
06.6 Instrucciones de uso y manipulación
Los medicamentos no utilizados y los desechos derivados de este medicamento deben eliminarse de acuerdo con las normativas locales.
07.0 TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Chiesi Farmaceutici S.p.A., Via Palermo 26 / A, Parma.
08.0 NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
CLENIL 100 mcg polvo para inhalación - AIC n. 023103106
CLENIL 200 mcg polvo para inhalación - AIC n. 023103118
CLENIL 400 mcg polvo para inhalación - AIC n. 023103120
09.0 FECHA DE LA PRIMERA AUTORIZACIÓN O RENOVACIÓN DE LA AUTORIZACIÓN
23 de diciembre de 1999
10.0 FECHA DE REVISIÓN DEL TEXTO
Diciembre 2012