Ingredientes activos: Sorafenib
Nexavar 200 mg comprimidos recubiertos con película.
¿Por qué se usa Nexavar? ¿Para qué sirve?
Nexavar se utiliza para el tratamiento del hepatocarcinoma.
Nexavar también se usa para tratar el cáncer de riñón (carcinoma de células renales avanzado) cuando se encuentra en una etapa avanzada y cuando la terapia estándar no ha ayudado a detenerlo o se considera inadecuado.
Nexavar se usa para tratar el cáncer de tiroides (cáncer de tiroides diferenciado).
Nexavar es un inhibidor de múltiples quinasas. Actúa reduciendo la velocidad de crecimiento de las células cancerosas y bloqueando el suministro de sangre que permite el crecimiento de las células cancerosas.
Contraindicaciones Cuándo no se debe usar Nexavar
No tome Nexavar
- si es alérgico al sorafenib oa cualquiera de los demás componentes de este medicamento (incluidos en la sección 6).
Precauciones de uso Lo que necesita saber antes de tomar Nexavar
Hable con su médico o farmacéutico antes de tomar Nexavar.
Tenga especial cuidado con Nexavar especialmente
- Si ocurren problemas en la piel. Nexavar puede provocar erupciones y reacciones cutáneas, especialmente en las manos y los pies. Por lo general, el médico puede tratar estos efectos. De lo contrario, el médico puede suspender el tratamiento o detenerlo por completo.
- Si tiene la presión arterial alta. Nexavar puede provocar un aumento de la presión arterial; Su médico controlará su presión arterial con regularidad y puede recetarle medicamentos para tratar la presión arterial alta.
- Si tiene problemas de sangrado o si está tomando warfarina o fenprocomona. El tratamiento con Nexavar puede aumentar el riesgo de hemorragia. Si está tomando warfarina o fenprocomona, medicamentos que diluyen la sangre para prevenir la formación de coágulos, puede haber un mayor riesgo de hemorragia.
- Si tiene dolores de pecho o problemas cardíacos. Su médico puede decidir interrumpir el tratamiento o interrumpirlo por completo.
- Si tiene un trastorno cardíaco, como una "alteración de la señal eléctrica llamada" prolongación del intervalo QT ".
- Si está a punto de someterse o acaba de someterse a una cirugía. Nexavar puede afectar la cicatrización de heridas. Si está a punto de someterse a una cirugía, probablemente se suspenderá su tratamiento con Nexavar. Luego, su médico decidirá cuándo retirarlo.
- Si está en tratamiento con irinotecán o docetaxel, que también son medicamentos para el cáncer, Nexavar puede aumentar los efectos y especialmente los efectos secundarios de estos medicamentos.
- Si está tomando neomicina u otros antibióticos. La eficacia de Nexavar puede disminuir: si tiene insuficiencia hepática grave, es posible que los efectos secundarios se agraven al tomar este medicamento.
- Si tiene la función renal reducida. Su médico controlará su equilibrio hídrico y electrolítico.
- Fertilidad. Nexavar puede reducir la fertilidad tanto en hombres como en mujeres. Si este es su caso, hable con su médico.
- Puede producirse una perforación gastrointestinal durante el tratamiento (ver sección 4: Posibles efectos adversos). En este caso, el médico interrumpirá el tratamiento.
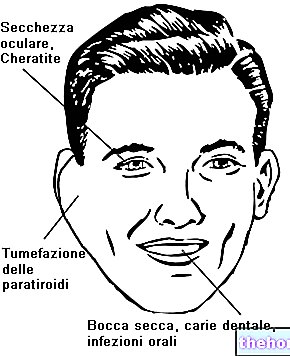
- Si tiene cáncer de tiroides, su médico controlará los niveles de calcio y hormona tiroidea en su sangre.
Informe a su médico si se encuentra en alguna de estas situaciones. Puede que necesite tratamiento para estos problemas, o su médico puede cambiar la dosis de Nexavar o interrumpir el tratamiento por completo (ver también la sección 4: Posibles efectos adversos).
Niños y adolescentes
Nexavar aún no se ha estudiado en niños y adolescentes.
Interacciones ¿Qué medicamentos o alimentos pueden cambiar el efecto de Nexavar?
Algunos medicamentos pueden afectar a Nexavar o verse afectados por él. Informe a su médico o farmacéutico si está tomando, ha tomado recientemente o podría tomar alguno de los medicamentos de esta lista o cualquier otro medicamento, incluso los adquiridos sin receta:
- Rifampicina, neomicina u otros medicamentos utilizados para tratar infecciones (antibióticos).
- Hypericum perforatum, también conocido como "hierba de San Juan", un tratamiento a base de hierbas para la depresión.
- Fenitoína, carbamazepina o fenobarbital, tratamientos para la epilepsia y otras enfermedades.
- Dexametasona, un corticosteroide que se usa para diversas enfermedades.
- Warfarina o fenprocomona, anticoagulantes utilizados para prevenir la formación de coágulos de sangre
- Doxorrubicina, capecitabina, docetaxel, paclitaxel e irinotecán, utilizados en el tratamiento del cáncer.
- Digoxina, utilizada en el tratamiento de la insuficiencia cardíaca leve o moderada.
Advertencias Es importante saber que:
Embarazo y lactancia
Evite quedar embarazada mientras recibe tratamiento con Nexavar. Si está en edad fértil, debe utilizar un método anticonceptivo eficaz durante el tratamiento con Nexavar. Si queda embarazada mientras está en tratamiento con Nexavar, informe a su médico de inmediato, quien decidirá si el tratamiento debe continuarse o interrumpirse.
No debe amamantar a su bebé mientras esté en tratamiento con Nexavar, ya que este medicamento puede interferir con el crecimiento y desarrollo de su bebé.
Conducción y uso de máquinas
No hay motivos para creer que Nexavar afectará a la capacidad para conducir o utilizar máquinas.
Dosis, método y momento de administración Cómo usar Nexavar: Posología
La dosis recomendada de Nexavar para adultos es de dos comprimidos de 200 mg dos veces al día.
Estos corresponden a una dosis diaria de 800 mg, o cuatro comprimidos al día. Tome los comprimidos de Nexavar con un vaso de agua, entre comidas o con alimentos bajos o medianos en grasa No tome este medicamento con alimentos muy grasos, ya que pueden reducir su eficacia. Si planea comer alimentos muy grasos, tome las tabletas al menos 1 hora antes o 2 horas después del almuerzo. Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte a su médico o farmacéutico.
Es importante tomar este medicamento aproximadamente a la misma hora todos los días para mantener constante la concentración en sangre.
Este medicamento generalmente se toma siempre que se observen beneficios clínicos y no haya efectos secundarios intolerables.
Sobredosis Qué hacer si ha tomado demasiado Nexavar
Si toma más Nexavar del que debiera
Informe a su médico de inmediato si usted o cualquier otra persona ha tomado más de la dosis recetada. Tomar demasiado Nexavar hace que los efectos secundarios sean más probables o más graves, especialmente diarrea y reacciones cutáneas. Es posible que su médico le indique que deje de tomar este medicamento.
Si olvidó tomar Nexavar
Si olvidó tomar una dosis, tómela tan pronto como se acuerde. Si su próxima dosis es poco tiempo, olvide la dosis omitida y continúe con su frecuencia
Efectos secundarios ¿Cuáles son los efectos secundarios de Nexavar?
Como todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran. Este medicamento también puede alterar los resultados de algunos análisis de sangre.
Muy común:
puede afectar a más de 1 de cada 10 personas
- Diarrea
- malestar (náuseas)
- sentirse débil o cansado (fatiga)
- dolor (incluyendo dolor en la boca, abdomen, dolor de cabeza, dolor de huesos, dolor por cáncer)
- pérdida de cabello (alopecia)
- enrojecimiento o dolor en las palmas de las manos o las plantas de los pies (reacción cutánea mano-pie)
- picazón o sarpullido
- Él vomitó
- hemorragia (incluida hemorragia en el cerebro, la pared intestinal y el tracto respiratorio)
- presión arterial alta o aumento de la presión arterial (hipertensión)
- infecciones
- pérdida de apetito (anorexia)
- estreñimiento
- dolor articular (artralgia)
- fiebre
- pérdida de peso
- sequedad de la piel
Común:
puede afectar hasta 1 de cada 10 personas
- enfermedad similar a la gripe
- indigestión (dispepsia)
- dificultad para tragar (disfagia)
- inflamación o sequedad de la boca, dolor en la lengua (estomatitis e inflamación de la membrana mucosa)
- niveles bajos de calcio en sangre (hipocalcemia)
- niveles bajos de potasio en sangre (hipopotasemia)
- dolor muscular (mialgia)
- trastornos de la sensibilidad en los dedos de las manos y los pies, que incluyen hormigueo y entumecimiento (neuropatía sensorial periférica)
- depresión
- problemas de erección (impotencia)
- cambios de voz (disfonía)
- acné
- piel inflamada, seca o descamada (dermatitis, descamación de la piel)
- insuficiencia cardiaca
- ataque cardíaco (infarto de miocardio) o dolor en el pecho
- tinnitus (zumbido en los oídos)
- insuficiencia renal
- altos niveles de proteína en la orina (proteinuria)
- debilidad general o pérdida de fuerza (astenia)
- número reducido de glóbulos blancos (leucopenia y neutropenia)
- número reducido de glóbulos rojos (anemia)
- bajo número de plaquetas en la sangre (trombocitopenia)
- inflamación de los folículos pilosos (foliculitis)
- actividad tiroidea reducida (hipotiroidismo)
- niveles bajos de sodio en sangre (hiponatremia)
- cambios en el sentido del gusto (disgeusia)
- enrojecimiento de la cara y, a menudo, otras áreas de la piel (enrojecimiento)
- secreción nasal (secreción nasal)
- acidez (enfermedad por reflujo gastroesofágico)
- cáncer de piel (queratoacantoma / cáncer de piel de células escamosas)
- engrosamiento de la capa externa de la piel (hiperqueratosis)
- contracción involuntaria repentina de un músculo (espasmos musculares)
Poco común:
puede afectar hasta 1 de cada 100 personas
- inflamación del estómago (gastritis)
- dolor de estómago (abdomen) debido a pancreatitis, inflamación de la vesícula biliar y / o conductos biliares
- coloración amarillenta de la piel o los ojos (ictericia) causada por niveles altos de pigmentos biliares (hiperbilirrubinemia)
- reacciones de tipo alérgico (incluidas reacciones cutáneas y urticaria)
- deshidración
- agrandamiento de los senos (ginecomastia)
- dificultad para respirar (enfermedad pulmonar)
- eczema
- actividad tiroidea excesiva (hipertiroidismo)
- múltiples erupciones cutáneas (eritema multiforme)
- Alta presión sanguínea
- perforación gastrointestinal
- edema reversible en la parte posterior del cerebro que puede estar asociado con dolor de cabeza, alteración de la conciencia, convulsiones y síntomas visuales, incluida la pérdida de la visión (leucoencefalopatía posterior reversible)
- reacción alérgica repentina y grave (reacción anafiláctica)
Raro:
puede afectar hasta 1 de cada 1.000 personas
- reacción alérgica con hinchazón de la piel (por ejemplo, cara, lengua) que puede causar dificultad para respirar y tragar (angioedema)
- ritmo cardíaco anormal (prolongación del intervalo QT)
- Inflamación del hígado, que puede provocar náuseas, vómitos, dolor abdominal e ictericia (hepatitis inducida por fármacos).
- una "erupción similar a una quemadura solar en la piel previamente expuesta a la radioterapia y que puede ser grave (dermatitis de tipo actínico)
- reacciones graves de la piel y / o membranas mucosas que pueden incluir ampollas dolorosas y fiebre, con desprendimiento de grandes áreas de piel (síndrome de Stevens-Johnson y necrólisis epidérmica tóxica)
- lesión muscular anormal que puede provocar problemas renales (rabdomiólisis)
- daño renal que provoca la pérdida de grandes cantidades de proteínas en la orina (síndrome nefrótico)
- inflamación de los vasos sanguíneos de la piel que puede manifestarse como una erupción (vasculitis leucocitoclástica)
No conocida:
la frecuencia no puede estimarse a partir de los datos disponibles
- deterioro de la función cerebral que puede estar asociado, por ejemplo, con somnolencia, cambios de comportamiento o confusión (encefalopatía)
Notificación de efectos secundarios
Si experimenta cualquier efecto adverso, consulte a su médico o farmacéutico, incluido cualquier posible efecto adverso no mencionado en este prospecto. También puede notificar los efectos secundarios directamente a través del sistema de notificación nacional que figura en el Apéndice V. Al notificar los efectos secundarios, puede ayudar a proporcionar más información sobre la seguridad de este medicamento.
Caducidad y retención
Mantenga este medicamento fuera de la vista y del alcance de los niños.
No utilice este medicamento después de la fecha de caducidad que aparece en la caja después de CAD y en cada blíster después de CAD. La fecha de vencimiento se refiere al último día de ese mes.
No almacene este medicamento por encima de 25 ° C.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que no necesita, esto ayudará a proteger el medio ambiente.
¿Qué contiene Nexavar?
- El ingrediente activo es sorafenib. Cada comprimido recubierto con película contiene 200 mg de sorafenib (como tosilato).
- Los demás componentes son: Núcleo del comprimido: croscarmelosa de sodio, celulosa microcristalina, hipromelosa, lauril sulfato de sodio y estearato de magnesio. Recubrimiento del comprimido: hipromelosa, macrogol, dióxido de titanio (E 171) y óxido de hierro rojo (E 172)
Aspecto de Nexavar y contenido del envase
Los comprimidos recubiertos con película de Nexavar 200 mg son rojos y redondos, con la cruz de Bayer en una cara y "200" en la otra, y se presentan en cajas de 112 comprimidos, que contienen cuatro blísteres calendario transparentes de 28 comprimidos cada uno.
Prospecto fuente: AIFA (Agencia Italiana de Medicamentos). Contenido publicado en enero de 2016. Es posible que la información presente no esté actualizada.
Para tener acceso a la versión más actualizada, es recomendable acceder al sitio web de la AIFA (Agencia Italiana de Medicamentos). Descargo de responsabilidad e información útil.
01.0 NOMBRE DEL MEDICAMENTO
NEXAVAR 200 MG
02.0 COMPOSICIÓN CUALITATIVA Y CUANTITATIVA
Cada comprimido recubierto con película contiene 200 mg de sorafenib (como tosilato).
Para consultar la lista completa de excipientes, ver sección 6.1.
03.0 FORMA FARMACÉUTICA
Comprimido recubierto con película (comprimido).
Comprimidos recubiertos con película de color rojo, redondos, biconvexos, marcados con una cruz de Bayer en una cara y "200" en la otra.
04.0 INFORMACIÓN CLÍNICA
04.1 Indicaciones terapéuticas
Hepatocarcinoma
Nexavar está indicado para el tratamiento del carcinoma hepatocelular (ver sección 5.1).
Carcinoma de células renales
Nexavar está indicado para el tratamiento de pacientes con carcinoma de células renales avanzado que no han respondido a la terapia previa con interferón alfa o interleucina-2, o que no se consideran elegibles para dicha terapia.
Cáncer de tiroides diferenciado
Nexavar está indicado para el tratamiento de pacientes con cáncer de tiroides diferenciado localmente avanzado o metastásico, progresivo, refractario al yodo radiactivo (papilar / folicular / de células de Hürthle).
04.2 Posología y forma de administración
El tratamiento con Nexavar debe estar bajo la supervisión de un médico con experiencia en el uso de terapias contra el cáncer.
Dosis
La dosis recomendada de Nexavar para adultos es de 400 mg de sorafenib (dos comprimidos de 200 mg) dos veces al día (equivalente a una dosis diaria total de 800 mg).
El tratamiento debe continuar mientras se observe un beneficio clínico o hasta que aparezcan toxicidades inaceptables.
Ajuste de la dosis
El manejo de las sospechas de reacciones adversas al medicamento puede requerir la interrupción temporal o la reducción de la dosis del tratamiento con sorafenib.
Cuando se requiere una reducción de la dosis durante el tratamiento del carcinoma hepatocelular (carcinoma hepatocelular, HCC) y carcinoma de células renales (Carcinoma de células renales, RCC), la dosis de Nexavar debe reducirse a dos comprimidos de sorafenib de 200 mg una vez al día (ver sección 4.4).
Cuando se requiere una reducción de la dosis durante el tratamiento del cáncer de tiroides diferenciado (carcinoma diferenciado de tiroides, DTC), la dosis de Nexavar debe reducirse a 600 mg de sorafenib al día en dosis divididas (dos comprimidos de 200 mg y un comprimido de 200 mg con un intervalo de doce horas).
Si se requiere una reducción adicional de la dosis, Nexavar puede reducirse a 400 mg de sorafenib al día en dosis divididas (dos comprimidos de 200 mg con doce horas de diferencia) y, si es necesario, reducirse a un comprimido de 200 mg una vez al día. reacciones adversas, se puede aumentar la dosis de Nexavar.
Población pediátrica
La seguridad y eficacia de Nexavar en niños mayores y adolescentes
Poblacion vieja
Para la población de edad avanzada (pacientes mayores de 65 años) no es necesario un ajuste de dosis.
Insuficiencia renal
No es necesario ajustar la dosis en pacientes con insuficiencia renal leve, moderada o grave. No hay datos disponibles para pacientes en diálisis (ver sección 5.2).
Se recomienda la monitorización del equilibrio hídrico y electrolítico en pacientes con riesgo de insuficiencia renal.
Deterioro hepático
No se requiere ajuste de dosis para pacientes con insuficiencia hepática Child Pugh A o B (leve a moderada). No se dispone de datos sobre pacientes con insuficiencia hepática Child Pugh C grave (ver secciones 4.4 y 5.2).
Método de administración
Para uso oral
El sorafenib debe administrarse entre comidas o con una comida baja o moderada en grasas. Si el paciente tiene la intención de ingerir una comida rica en grasas, los comprimidos de sorafenib deben tomarse al menos una hora antes o dos horas después de la comida, tragándose los comprimidos con un vaso de agua.
04.3 Contraindicaciones
Hipersensibilidad al principio activo oa alguno de los excipientes incluidos en la sección 6.1.
04.4 Advertencias especiales y precauciones de uso apropiadas
Toxicidad dermatológica
Reacción cutánea mano-pie (eritrodisestesia palmoplantar) e sarpullido representan las reacciones adversas más frecuentes al sorafenib. Sarpullido y la reacción cutánea mano-pie suelen ser de Grado 1 y 2, según i Criterios de toxicidad comunes (CTC), y generalmente aparecen durante las primeras seis semanas de tratamiento con sorafenib. El manejo de la toxicidad dermatológica puede incluir terapias tópicas para aliviar los síntomas, interrupción temporal del tratamiento y / o un cambio en la dosis de sorafenib, o en casos severos o persistentes, interrupción definitiva de su administración (ver sección 4.8).
Hipertensión
Se observó una mayor incidencia de hipertensión arterial en los pacientes tratados con sorafenib. En estos pacientes, la hipertensión fue generalmente de leve a moderada, se presentó en las primeras etapas del tratamiento y respondió al tratamiento antihipertensivo estándar. La presión arterial debe controlarse regularmente y tratarse según sea necesario de acuerdo con la práctica médica actual. En caso de hipertensión grave o persistente, o una crisis hipertensiva, a pesar del inicio del tratamiento antihipertensivo, se recomienda considerar la suspensión permanente de la administración de sorafenib (ver sección 4.8).
Hemorragia
El riesgo de hemorragia puede aumentar tras la administración de sorafenib. Si un episodio hemorrágico requiere intervención médica, se recomienda considerar la interrupción permanente de la administración de sorafenib (ver sección 4.8).
Isquemia cardíaca y / o infarto
En un estudio doble ciego, aleatorizado y controlado con placebo (estudio 1, ver sección 5.1), la incidencia de infarto cardíaco o isquemia al inicio del tratamiento fue mayor en el grupo de sorafenib (4,9%) que en el grupo de tratamiento con placebo (0,4%). En el estudio 3 (ver sección 5.1), la incidencia de infarto de miocardio o isquemia al inicio del tratamiento fue del 2,7% en los pacientes tratados con sorafenib y del 1,3% en los pacientes tratados con placebo. Los pacientes con arteriopatía coronaria inestable o con infarto de miocardio reciente fueron excluidos de estos estudios. Se debe considerar la necesidad de la interrupción temporal o permanente del tratamiento con sorafenib en pacientes que desarrollen isquemia cardíaca y / o infarto (ver sección 4.8).
Prolongación del intervalo QT
Se ha demostrado que sorafenib prolonga el intervalo QT / QTc (ver sección 5.1), lo que puede conducir a un mayor riesgo de arritmia ventricular. Use sorafenib con precaución en pacientes que tienen o pueden desarrollar una prolongación del QTc, como los pacientes con QT largo congénito. Síndrome, aquellos tratados con una dosis acumulada alta de antraciclinas, pacientes que toman ciertos fármacos antiarrítmicos u otros fármacos que pueden provocar una prolongación del intervalo QT y aquellos con alteraciones electrolíticas, por ejemplo hipopotasemia, hipocalcemia o hipomagnesemia. Cuando se utiliza sorafenib en estos pacientes, electrocardiografía periódica y las mediciones de electrolitos (magnesio, potasio y calcio) deben realizarse durante el período de tratamiento.
Perforación gastrointestinal
La perforación gastrointestinal es un evento poco común y se ha notificado en menos del 1% de los pacientes que toman sorafenib. En algunos casos, no hubo asociación con un tumor intraabdominal obvio. En caso de perforación gastrointestinal, debe interrumpirse la administración de sorafenib (ver sección 4.8).
Deterioro hepático
No se dispone de datos en pacientes con insuficiencia hepática grave (Child Pugh C). En estos pacientes, la exposición puede aumentar ya que sorafenib se elimina principalmente a través del hígado (ver secciones 4.2 y 5.2).
Administración concomitante de warfarina
Episodios hemorrágicos poco frecuentes o aumento del INR (Internacional normalizado
Proporción) en algunos pacientes que tomaban warfarina durante el tratamiento con sorafenib. Los pacientes en tratamiento con warfarina o fenprocumón deben ser controlados regularmente para detectar cambios en el tiempo de protrombina, INR o episodios hemorrágicos clínicamente relevantes (ver secciones 4.5 y 4.8).
Complicaciones en la cicatrización de heridas.
No se han realizado estudios formales sobre el efecto de sorafenib en la cicatrización de heridas. Se recomienda la suspensión temporal del tratamiento con sorafenib por razones de precaución en pacientes sometidos a cirugía mayor. La experiencia clínica sobre cuándo reiniciar el tratamiento después de una cirugía mayor es limitada. Por lo tanto, la decisión de reanudar el tratamiento con sorafenib después de una cirugía mayor debe basarse en una evaluación clínica de la cicatrización adecuada de la herida.
Poblacion vieja
Se han notificado casos de insuficiencia renal. Por tanto, se debe considerar la monitorización de la función renal.
Interacción entre drogas
Se recomienda precaución al administrar sorafenib con sustancias que se metabolizan y / o eliminan predominantemente a través de las vías UGT1A1 (p. Ej., Irinotecán) o UGT1A9 (ver sección 4.5).
Se recomienda precaución en caso de administración concomitante de sorafenib y docetaxel (ver sección 4.5).
La combinación con neomicina o con otros antibióticos capaces de causar graves alteraciones ecológicas en la microflora gastrointestinal puede conducir a una disminución de la biodisponibilidad de sorafenib (ver sección 4.5). Se debe evaluar el riesgo de disminución de la concentración plasmática de sorafenib antes de comenzar. un curso de tratamiento con antibióticos.
Se observó una mayor mortalidad en pacientes con cáncer de pulmón de células escamosas tratados con sorafenib en combinación con quimioterapia a base de platino.
En dos ensayos clínicos aleatorizados, que estudiaron a pacientes con cáncer de pulmón de células no pequeñas (Cáncer de pulmón de células no pequeñas, CPCNP), el cociente de riesgo (HR) para la supervivencia global en un subgrupo de pacientes con cáncer de pulmón de células escamosas fue de 1,81 (IC del 95%: 1,19; 2,74) en los pacientes tratados con sorafenib además del tratamiento con paclitaxel / carboplatino y 1,22 (95% IC 0,82; 1,80) en pacientes tratados con sorafenib además de la terapia con gemcitabina / cisplatino. No se observó una causa predominante de muerte, pero se observó una mayor incidencia de insuficiencia respiratoria, hemorragia e infección en pacientes tratados con sorafenib además de la terapia a base de platino.
Advertencias específicas de patología
Carcinoma diferenciado de tiroides (DTC)
Antes de iniciar el tratamiento, se recomienda a los médicos que evalúen cuidadosamente el pronóstico de cada paciente en función del tamaño máximo de la lesión (ver sección 5.1), los síntomas relacionados con la enfermedad (ver sección 5.1) y la velocidad de progresión.
El tratamiento de las sospechas de reacciones adversas al medicamento puede requerir una "interrupción temporal o una reducción de la dosis del tratamiento con sorafenib. En el estudio 5 (ver sección 5.1), el 37% de los sujetos interrumpió temporalmente el tratamiento y el 35% redujo la dosis tan pronto como el ciclo 1 del tratamiento con sorafenib".
La reducción de la dosis fue sólo parcialmente eficaz para aliviar las reacciones adversas, por lo que se recomienda realizar evaluaciones repetidas de los riesgos y los beneficios, teniendo en cuenta la actividad antitumoral y la tolerabilidad.
Hemorragia en el DTC
Debido al riesgo potencial de hemorragia, la infiltración traqueal, bronquial y esofágica debe tratarse con terapia local antes de administrar sorafenib a pacientes con CDT.
Hipocalcemia en el DTC
Cuando se usa sorafenib en pacientes con CDT, se recomienda una estrecha monitorización de los niveles de calcio en sangre. En los ensayos clínicos, la hipocalcemia fue más frecuente y más grave en pacientes con CDT, especialmente en aquellos con antecedentes de hipoparatiroidismo, en comparación con pacientes con carcinoma de células renales o hepatocarcinoma. Se produjo hipocalcemia de grado 3 y 4. Se manifestó en el 6,8% y el 3,4% de los casos. Pacientes con CDT tratados con sorafenib (ver sección 4.8). La hipocalcemia grave debe corregirse para prevenir complicaciones como la prolongación del intervalo QT o torsades de pointes (ver la sección de prolongación del intervalo QT).
Supresión de TSH en el DTC
En el estudio 5 (ver sección 5.1), se observaron aumentos en los niveles de TSH superiores a 0,5 mU / L en pacientes tratados con sorafenib. Se recomienda un estrecho seguimiento de los niveles de TSH cuando se utiliza sorafenib en pacientes con CDT.
Carcinoma de células renales
Pacientes de alto riesgo, según la definición del grupo pronóstico de MSKCC (Centro Oncológico Memorial Sloan Kettering), no se incluyeron en el ensayo clínico de fase III en carcinoma de células renales (ver estudio 1 en la sección 5.1) y no se ha establecido la relación beneficio-riesgo en estos pacientes.
04.5 Interacciones con otros medicamentos y otras formas de interacción
Inductores de enzimas metabólicas.
La administración de rifampicina durante 5 días antes de la administración de una dosis única de sorafenib resultó en una reducción media del AUC de sorafenib del 37% .Otros inductores de CYP3A4 y / o glucuronidación (p. Ej. Hypericum perforatum también conocida como "hierba de San Juan", fenitoína, carbamazepina, fenobarbital y dexametasona) pueden aumentar el metabolismo de sorafenib y reducir así su concentración.
Inhibidores de CYP3A4
El ketoconazol, un potente inhibidor del CYP3A4, administrado una vez al día durante 7 días a voluntarios varones sanos, no alteró el AUC medio de una dosis única de 50 mg de sorafenib. Estos datos sugieren que las interacciones farmacocinéticas clínicas de sorafenib con inhibidores del CYP3A4 son poco probables.
Sustratos CYP2B6, CYP2C8 y CYP2C9
In vitro sorafenib inhibe CYP2B6, CYP2C8 y CYP2C9 con una potencia casi igual. Sin embargo, en estudios de farmacocinética clínica, la coadministración de 400 mg de sorafenib dos veces al día con ciclofosfamida, un sustrato de CYP2B6, o paclitaxel, un sustrato de CYP2C8, no produjo una inhibición clínicamente significativa. Estos datos sugieren que sorafenib, a la dosis recomendada de 400 mg dos veces al día, puede no ser un inhibidor en vivo CYP2B6 o CYP2C8.
Además, el tratamiento concomitante con sorafenib y warfarina, un sustrato de CYP2C9, no produjo cambios en la media de PT-INR en comparación con placebo. Por lo tanto, también el riesgo de una "inhibición en vivo El CYP2C9 clínicamente relevante de sorafenib puede considerarse bajo. Sin embargo, los pacientes que toman warfarina o fenprocumón deben controlar regularmente su INR (ver sección 4.4).
Sustratos CYP3A4, CYP2D6 y CYP2C19
La administración concomitante de sorafenib y midazolam, dextrometorfano u omeprazol, que son sustratos de los citocromos CYP3A4, CYP2D6 y CYP2C19, respectivamente, no alteró la exposición a estos agentes, lo que indica que sorafenib no es ni un inhibidor ni un inductor de estas isoenzimas. citocromo P450, por lo que es improbable que se produzcan interacciones farmacocinéticas clínicas de sorafenib con sustratos de estas enzimas.
Sustratos UGT1A1 y UGT1A9
In vitro, sorafenib inhibió la glucuronidación por UGT1A1 y UGT1A9. Se desconoce la relevancia clínica de este hallazgo (ver más abajo y la sección 4.4).
Educación in vitro sobre la inducción de enzimas del sistema CYP
Las actividades de CYP1A2 y CYP3A4 no se alteraron después de la exposición de cultivos de hepatocitos humanos a sorafenib, lo que indica que es poco probable que sorafenib sea un inductor de CYP1A2 y CYP3A4.
Sustratos para P-gp
In vitro, se ha demostrado que sorafenib inhibe la proteína de transporte p-glicoproteína (P-gp). En caso de tratamiento concomitante con sorafenib, no se puede excluir un aumento en la concentración plasmática de sustratos para P-gp, como digoxina.
Asociación con otros agentes antineoplásicos
En estudios clínicos, sorafenib se administró con varios otros agentes antineoplásicos en su posología de uso común, incluyendo gemcitabina, cisplatino, oxaliplatino, paclitaxel, carboplatino, capecitabina, doxorrubicina, irinotecán, docetaxel y ciclofosfamida. Sorafenib no tuvo ningún efecto clínicamente relevante sobre la farmacocinética de gemcitabina, cisplatino, carboplatino, oxaliplatino o ciclofosfamida.
Paclitaxel / carboplatino
• Administración de paclitaxel (225 mg / m2) y carboplatino (AUC = 6) con sorafenib (≤ 400 mg dos veces al día), con una interrupción de 3 días de la administración de sorafenib (los dos días anteriores y el día de la administración de paclitaxel / carboplatino ), no tuvo un efecto significativo sobre la farmacocinética de paclitaxel.
• La administración concomitante de paclitaxel (225 mg / m2, una vez cada 3 semanas) y carboplatino (AUC = 6) con sorafenib (400 mg dos veces al día, sin interrumpir la dosificación de sorafenib) resultó en un aumento del 47% en la exposición a sorafenib, un 29% aumento en la exposición al paclitaxel y un aumento del 50% en la exposición al 6-OH paclitaxel La farmacocinética del carboplatino no se vio afectada.
Estos datos indican que no es necesario ajustar la dosis cuando se coadministra paclitaxel y carboplatino con sorafenib con una interrupción de 3 días de la administración de sorafenib (los dos días anteriores y el día de la administración de paclitaxel / carboplatino). aumento de la exposición a sorafenib y paclitaxel poco después de la coadministración de sorafenib sin interrupción de la dosis.
Capecitabina
La administración concomitante de capecitabina (750-1050 mg / m2 dos veces al día, días 1-14 cada 21 días) y sorafenib (200 o 400 mg dos veces al día sin interrupción de la dosificación) no produjo cambios significativos en la exposición. A sorafenib, pero un 15 -Aumento del 50% en la exposición a capecitabina y un aumento del 0-52% en la exposición al 5-FU Se desconoce la relevancia clínica de estos pequeños y modestos aumentos en la exposición al 5-FU, capecitabina y 5-FU cuando se coadministra con sorafenib.
Doxorrubicina / Irinotecán
El tratamiento concomitante con sorafenib resultó en un aumento del 21% en el AUC de doxorrubicina.Cuando se administró con irinotecán, cuyo metabolito SN-38 se metaboliza posteriormente a través de la vía UGT1A1, hubo un aumento del 67-120% en el AUC del SN-38 y un aumento del 26-42% en el AUC del irinotecán. Se desconoce la relevancia clínica de estos datos ( ver sección 4.4).
Docetaxel
Docetaxel (una dosis de 75 o 100 mg / m2 cada 21 días) administrado concomitantemente con sorafenib (200 mg o 400 mg dos veces al día en los días 2 a 19 de un ciclo de tratamiento de 21 días, con una interrupción de 3 días correspondiente a docetaxel administración) resultó en un aumento en el AUC y Cmax de docetaxel de 36 - 80% y 16 - 32%, respectivamente. Se recomienda precaución cuando se coadministra con sorafenib y docetaxel (ver sección 4.4).
Asociación con otros agentes
Neomicina
La combinación con neomicina, un agente antimicrobiano no sistémico utilizado para erradicar la flora gastrointestinal, interfiere con la recirculación enterohepática de sorafenib (ver sección 5.2, Biotransformación y metabolismo), lo que reduce la exposición a sorafenib. En voluntarios sanos tratados con neomicina durante 5 días, la exposición media a sorafenib disminuyó en un 54%. No se han estudiado los efectos de otros antibióticos, pero lo más probable es que dependan de su capacidad para interferir con los microorganismos con actividad glucuronidasa.
04.6 Embarazo y lactancia
El embarazo
No existen datos sobre el uso de sorafenib en mujeres embarazadas. Los estudios en animales han mostrado toxicidad reproductiva, incluidas malformaciones (ver sección 5.3). Se ha demostrado que sorafenib y sus metabolitos atraviesan la placenta en ratas. Y se espera que sorafenib cause efectos nocivos. Sorafenib no debe usarse durante el embarazo a menos que sea claramente necesario, y solo después de una cuidadosa consideración de las necesidades de la madre y el riesgo para el feto.
Las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces durante el tratamiento.
Hora de la comida
Se desconoce si sorafenib se excreta en la leche materna. En los animales, sorafenib y / o sus metabolitos se excretan en la leche. Dado que sorafenib puede afectar al crecimiento y desarrollo del recién nacido (ver sección 5.3), las mujeres deben interrumpir la lactancia durante el tratamiento con sorafenib.
Fertilidad
Los estudios en animales muestran que sorafenib puede afectar la fertilidad masculina y femenina (ver sección 5.3).
04.7 Efectos sobre la capacidad para conducir y utilizar máquinas
No se han realizado estudios sobre la capacidad para conducir o utilizar máquinas. No hay motivos para creer que sorafenib afecte a la capacidad para conducir o utilizar máquinas.
04.8 Efectos indeseables
Las reacciones adversas graves más importantes fueron isquemia e infarto de miocardio, perforación gastrointestinal, hepatitis por fármacos, hemorragia e hipertensión o crisis hipertensiva.
Las reacciones adversas más frecuentes fueron diarrea, astenia, alopecia, infección, reacción cutánea mano-pie (correspondiente en MedDRA al "síndrome de eritrodisestesia palmo-plantar") y sarpullido.
Las reacciones adversas notificadas en diferentes ensayos clínicos o en el uso poscomercialización se enumeran en la tabla 1, ordenadas por MedDRA y frecuencia. Las frecuencias se definen de la siguiente manera: muy frecuentes (≥1 / 10), frecuentes (≥1 / 100,
Dentro de cada clase de frecuencia, las reacciones adversas se presentan en orden descendente de gravedad.
Tabla 1: Reacciones adversas globales notificadas en pacientes en diferentes estudios clínicos o en el uso poscomercialización.
* Las reacciones adversas pueden poner en peligro la vida o ser mortales. Estos eventos son poco comunes o menos frecuentes que infrecuentes.
** La reacción cutánea mano-pie corresponde al síndrome de eritrodisestesia palmoplantar en MedDRA
Obtenga más información sobre algunas reacciones adversas
Insuficiencia cardíaca congestiva
En un estudio clínico patrocinado por la empresa, se notificó insuficiencia cardíaca congestiva como evento adverso en el 1,9% de los pacientes tratados con sorafenib (N = 2276). En el estudio 11213 (RCC) se notificaron eventos adversos compatibles con insuficiencia cardíaca congestiva en el 1,7% de los pacientes tratados con sorafenib y en el 0,7% de los pacientes tratados con placebo. En el estudio 100554 (HCC), estos acontecimientos se notificaron en el 0,99% de los pacientes tratados con sorafenib y en el 1,1% de los pacientes tratados con placebo.
Información adicional para poblaciones especiales
En los estudios clínicos, ciertas reacciones adversas al fármaco, como reacciones cutáneas de manos y pies, diarrea, alopecia, pérdida de peso, hipertensión, hipocalcemia y queratoacantoma / carcinoma de piel de células escamosas, se produjeron con una frecuencia considerablemente mayor en pacientes con cáncer de tiroides diferenciado en comparación con los pacientes. incluido en los estudios de carcinoma de células renales o hepatocelulares.
Cambios en las pruebas de laboratorio en pacientes con CHC (estudio 3) y CCR (estudio 1)
Se ha informado con mucha frecuencia un aumento de la lipasa y la amilasa. Un aumento de grado 3 o 4 de la lipasa Criterios de toxicidad comunesLos eventos adversos (CTCAE) se produjeron en el 11% y el 9% de los pacientes del grupo de sorafenib en el Estudio 1 (RCC) y el Estudio 3 (HCC), respectivamente, frente al 7% y el 9% de los pacientes del grupo de sorafenib tratados con placebo. Se produjo una elevación de la amilasa CTCAE de grado 3 o 4 en el 1% y el 2% de los pacientes del grupo de sorafenib en el estudio 1 y el estudio 3, respectivamente, frente al 3% de los pacientes de ambos grupos de placebo. Se notificó pancreatitis clínica en 2 de 451 pacientes tratados con sorafenib (CTCAE Grado 4) en el Estudio 1, en 1 de 297 pacientes tratados con sorafenib (CTCAE Grado 2) en el Estudio 3 y en 1 de 451 pacientes (CTCAE Grado 2) tratados con placebo en el Estudio 1.
La hipofosfatemia es un hallazgo de laboratorio muy común y se observó en el 45% y el 35% de los pacientes tratados con sorafenib en el estudio 1 y el estudio 3, respectivamente, frente al 12% y el 11% de los pacientes tratados con placebo, respectivamente. En el ensayo 1 se produjo hipofosfatemia de grado 3 según CTCAE (1-2 mg / dl) en el 13% de los pacientes tratados con sorafenib y en el 3% de los pacientes tratados con placebo, mientras que en el ensayo 3 ocurrió en el 11% de los pacientes tratados con sorafenib y en 2% de los pacientes tratados con placebo No se han notificado casos de hipofosfatemia de grado 4 según CTCAE (se desconoce la etiología de la hipofosfatemia asociada con sorafenib).
Se observaron anomalías de laboratorio de grado 3 o 4 del CTCAE, incluidas linfopenia y neutropenia, en ≥ 5% de los pacientes tratados con sorafenib.
Se observó hipocalcemia en el 12% y el 26,5% de los pacientes tratados con sorafenib en comparación con el 7,5% y el 14,8% de los pacientes del grupo placebo en el estudio 1 y el estudio 3, respectivamente.Los casos de hipocalcemia fueron leves (CTCAE grados 1 y 2). A Se produjo hipocalcemia de grado 3 según CTCAE (6,0 - 7,0 mg / dl) en el 1,1% y el 1,8% de los pacientes tratados con sorafenib y en el 0,2% y el 1,1% de los pacientes del grupo placebo, y una hipocalcemia de grado 4 según CTCAE (
En los estudios 1 y 3, se observó una reducción del potasio en el 5,4% y el 9,5% de los pacientes tratados con sorafenib, respectivamente, en comparación con el 0,7% y el 5,9% de los pacientes que recibieron placebo. La mayoría de los casos de hipopotasemia fueron leves (CTCAE grado 1). En estos estudios, se produjo hipopotasemia CTCAE grado 3 en el 1,1% y el 0,4% de los pacientes tratados con sorafenib y en el 0,2% y 0,7% de los pacientes del grupo placebo.
Cambios en las pruebas de laboratorio en pacientes con CDT (estudio 5)
Se observó hipocalcemia en el 35,7% de los pacientes tratados con sorafenib en comparación con el 11,0% de los pacientes del grupo placebo. La mayoría de los casos de hipocalcemia fueron de gravedad leve. Se produjo hipocalcemia CTCAE grado 3 en el 6,8% de los pacientes tratados con sorafenib y en el 1,9% de los pacientes del grupo placebo, mientras que la hipocalcemia CTCAE grado 4 ocurrió en el 3,4% de los pacientes, los pacientes tratados con sorafenib y el 1,0% de los pacientes del grupo placebo.
Otros cambios de laboratorio clínicamente relevantes observados en el estudio 5 se muestran en la Tabla 2.
Tabla 2: Anomalías de laboratorio emergentes del tratamiento notificadas en pacientes con CDT (estudio 5) en la fase doble ciego
* Criterios terminológicos comunes para eventos adversos (CTCAE), versión 3.0
** Se desconoce la etiología de la hipofosfatemia asociada a sorafenib.
Notificación de sospechas de reacciones adversas
La notificación de sospechas de reacciones adversas que se produzcan después de la autorización del medicamento es importante, ya que permite un seguimiento continuo de la relación beneficio / riesgo del medicamento. Se ruega a los profesionales sanitarios que notifiquen cualquier sospecha de reacciones adversas a través del sitio web de la Agencia Italiana de Medicamentos. : www.agenziafarmaco.gov.it/it/responsabili.
04.9 Sobredosis
No existen tratamientos específicos en caso de sobredosis de sorafenib. La dosis más alta de sorafenib estudiada clínicamente es de 800 mg dos veces al día. Los eventos adversos observados después de esta dosis fueron principalmente diarrea y reacciones dermatológicas. Si se sospecha una sobredosis, se debe suspender el sorafenib y, si es necesario, iniciar la terapia de apoyo.
05.0 PROPIEDADES FARMACOLÓGICAS
05.1 Propiedades farmacodinámicas
Grupo farmacoterapéutico: agentes antineoplásicos, inhibidores de la proteína quinasa.
Código ATC: L01XE05.
El sorafenib es un inhibidor de la quinasa que ha demostrado propiedades tanto antiproliferativas como antiangiogénicas. in vitro y en vivo.
Mecanismo de acción y efectos farmacodinámicos.
Sorafenib es un inhibidor de la quinasa que inhibe la proliferación de células cancerosas. in vitro. Sorafenib inhibe el crecimiento de un amplio espectro de tumores humanos trasplantados en ratones atímicos, lo que también resulta en una reducción de la angiogénesis tumoral. Sorafenib inhibe la actividad de los objetivos presentes en la célula tumoral (CRAF, BRAF, V600E BRAF, c-KIT y FLT- 3) y en los vasos sanguíneos del tumor (CRAF, VEGFR-2, VEGFR-3 y PDGFR-ß). Las RAF quinasas son serina / treonina quinasas, mientras que c-KIT, FLT-3, VEGFR-2, VEGFR-3 y PDGFR-ß son tirosina quinasas receptoras.
Eficacia clínica
Se ha estudiado la seguridad y eficacia clínica de sorafenib en pacientes con carcinoma hepatocelular (carcinoma hepatocelular, HCC), en pacientes con carcinoma de células renales avanzado (Carcinoma de células renales, CCR) y en pacientes con cáncer de tiroides diferenciado (carcinoma diferenciado de tiroides, DTC).
Hepatocarcinoma
El estudio 3 (estudio 100554) fue un estudio clínico internacional de fase III, multicéntrico, aleatorizado, doble ciego, controlado con placebo, en 602 pacientes con cáncer hepatocelular. Las características demográficas y de la enfermedad basales fueron comparables entre los grupos de sorafenib y placebo con respecto a la clasificación del Grupo de Oncología Cooperativa Estern (ECOG) (grado 0: 54% versus 54%; grado 1: 38% versus 39%; grado 2: 8% versus 7%), a la clasificación TNM (estadio I:
El estudio se cerró después de que un análisis de supervivencia global (SG) provisional planificado excediera el límite de eficacia predefinido. Este análisis de SG mostró un aumento estadísticamente significativo en la SG para los pacientes tratados con sorafenib en comparación con los pacientes tratados con placebo (HR: 0,69, p = 0,00058, ver tabla 3).
En este estudio, los datos en pacientes con insuficiencia hepática Child Pugh B son limitados y solo se incluyó a un paciente Child Pugh C.
Tabla 3: Resultados de eficacia del estudio 3 (estudio 100554) en hepatocarcinoma
IC = intervalo de confianza, HR = cociente de riesgo (sorafenib sobre placebo)
* estadísticamente significativo ya que el valor p estaba por debajo del límite de corte predeterminado O ", establecido en 0,0077
** revisión radiológica independiente
Un segundo estudio de fase III, internacional, multicéntrico, aleatorizado, doble ciego, controlado con placebo (Estudio 4, 11849) evaluó el beneficio clínico de sorafenib en 226 pacientes con cáncer de hígado avanzado. Este estudio, realizado en China, Corea y Taiwán, confirmó los resultados del Estudio 3 con respecto al perfil beneficio-riesgo favorable de sorafenib (HR (SG): 0,68, p = 0,01414).
En los factores de estratificación predefinidos (clasificación ECOG, presencia o ausencia de invasión vascular macroscópica y / o diseminación extrahepática del tumor) de los Estudios 3 y 4, el HR estuvo consistentemente a favor del sorafenib sobre el placebo. Los análisis exploratorios de subgrupos sugirieron un efecto del tratamiento menos pronunciado en pacientes con metástasis distantes ya al inicio del estudio.
Carcinoma de células renales
La tolerabilidad y eficacia de sorafenib en el tratamiento del carcinoma de células renales avanzado (CCR) se estudiaron en dos estudios clínicos:
El estudio 1 (estudio 11213) fue un estudio clínico de fase III multicéntrico, aleatorizado, doble ciego y controlado con placebo en 903 pacientes. Solo se incluyeron pacientes con tumores renales de células claras y con factor de riesgo bajo y medio según MSKCC. los punto finalprimarios fueron la supervivencia general (SG, en general supervivencia) y supervivencia libre de progresión (SLP, Progresión Gratis Supervivencia).
Aproximadamente la mitad de los pacientes tenían su estado general igual a 0 en la escala ECOG y la mitad de los pacientes pertenecían al grupo pronóstico con una puntuación baja según la clasificación del MSKCC.
La SLP se evaluó de acuerdo con los criterios RECIST con una revisión radiológica independiente ciega. El análisis de SLP se realizó en 342 eventos en 769 pacientes. La mediana del valor de SLP fue de 167 días en los pacientes tratados con sorafenib en comparación con 84 días en los pacientes que recibieron placebo (HR = 0,44; IC del 95%: 0,35 - 0,55; p
Un análisis provisional (segundo análisis provisional) para la supervivencia global (en general supervivencia) se realizó en 367 muertes en 903 pacientes. El valor alfa nominal para este análisis fue 0,0094. La mediana de supervivencia fue de 19,3 meses en los pacientes tratados con sorafenib, en comparación con 15,9 meses en los pacientes aleatorizados a placebo (HR = 0,77; IC del 95%: 0,63-0,95; p = 0,015). En el momento del análisis, aproximadamente 200 pacientes pasaron del grupo de placebo al grupo de sorafenib.
El estudio 2 fue un estudio de fase II con suspensión aleatorizada del tratamiento en pacientes con cáncer metastásico, incluido el CCR. Los pacientes con enfermedad estable y en tratamiento con sorafenib fueron aleatorizados para recibir placebo o continuar con el tratamiento con sorafenib. La SSP en los pacientes con CCR fue significativamente mayor (163 días) para los pacientes tratados con sorafenib que el observado en los pacientes que recibieron placebo (41 días) (p = 0,0001, HR = 0,29).
Carcinoma diferenciado de tiroides (CDT)
El estudio 5 (estudio 14295) fue un estudio de fase III internacional, multicéntrico, aleatorizado, doble ciego, controlado con placebo, realizado en 417 pacientes con CDT localmente avanzado o refractario al yodo radiactivo metastásico. La supervivencia libre de progresión (SLP), determinada mediante una evaluación radiológica independiente ciega basada en los criterios RECIST, fue el criterio de valoración principal del estudio. Los criterios de valoración secundarios incluyeron la supervivencia global (SG), la tasa de respuesta tumoral y la duración de la respuesta. recibir sorafenib de etiqueta abierta.
Los pacientes se incluyeron en el estudio si progresaron dentro de los 14 meses anteriores a la inscripción y si tenían un CDT refractario al yodo radiactivo (yodo radiactivo, RAI). El CDT refractario a RAI se definió como la presencia de una lesión que no realza con yodo en la gammagrafía con RAI, o la administración acumulada de RAI ≥ 22,2 GBq, o la progresión después del tratamiento con RAI dentro de los 16 meses anteriores. una distancia máxima de 16 meses entre sí.
La demografía inicial y las características de los pacientes estaban bien equilibradas en los dos grupos de tratamiento. Las metástasis estaban presentes en los pulmones en el 86%, en los ganglios linfáticos en el 51% y en el hueso en el 27% de los pacientes. La mediana de la actividad acumulada de yodo radiactivo administrada antes de la inscripción fue de aproximadamente 14,8 GBq. La mayoría de los pacientes tenían carcinoma papilar (56,8%), seguido de carcinoma folicular (25,4%) y carcinoma poco diferenciado (9,6%).
La mediana de tiempo hasta la SLP fue de 10,8 meses en el grupo de sorafenib, en comparación con 5,8 meses en el grupo de placebo. (HR = 0,587; intervalo de confianza (IC) del 95%: 0,454, 0,758; p unilateral
El efecto de sorafenib sobre la SLP fue constante independientemente de la región geográfica, la edad por encima o por debajo de los 60 años, el sexo, la histología y la presencia o ausencia de metástasis óseas.
En un análisis de supervivencia global realizado 9 meses después de la fecha de corte para el análisis final de SLP, no hubo diferencias estadísticamente significativas en la supervivencia global entre los grupos de tratamiento (HR 0,884; IC 95%: 0,633; 1,236, p unilateral valor de 0,236). La mediana de SG no se logró en el grupo de sorafenib mientras que fue de 36,5 meses en el grupo de placebo.Ciento cincuenta y siete pacientes (75%) aleatorizados a placebo y 61 pacientes (30%) aleatorizados a sorafenib recibieron sorafenib abierto.
La duración media de la terapia en la fase doble ciego fue de 46 semanas (rango 0,3-135) para los pacientes que recibieron sorafenib y de 28 semanas (rango 1,7-132) para los pacientes que recibieron placebo.
No se observó una respuesta completa (respuesta completa, CR) según los criterios RECIST. La tasa de respuesta global (RC + respuesta parcial, respuesta parcial (PR)), determinada por evaluación radiológica independiente, fue mayor en el grupo de sorafenib (24 pacientes, 12,2%) en comparación con el grupo de placebo (1 paciente, 0,5%), p unilateral
Un análisis posterior a posterior de los subgrupos basado en el tamaño máximo del tumor mostró un efecto del tratamiento sobre la SLP a favor de sorafenib en comparación con placebo en pacientes con un tamaño máximo de lesión tumoral de 1,5 cm o más (HR 0,54 (IC del 95%: 0,41-0,71)) , mientras que se registró un efecto numéricamente menor en pacientes con tamaño máximo de lesión tumoral menor de 1,5 cm (HR 0,87 (IC 95%: 0,40 -1,89)).
Un análisis post-hoc basado en los síntomas relacionados con el cáncer de tiroides presentes en las condiciones basales mostró un efecto del tratamiento sobre la SLP a favor de sorafenib sobre el placebo en pacientes sintomáticos y asintomáticos. El valor de HR para la progresión de la supervivencia libre fue de 0,39 (IC del 95%: 0,21 - 0,72) para pacientes con síntomas basales y 0,60 (IC del 95%: 0,45 - 0,81) para pacientes sin síntomas en condiciones basales.
Prolongación del intervalo QT
En un estudio de farmacología clínica, se midió QT / QTc en 31 pacientes al inicio del tratamiento (antes del tratamiento) y después del tratamiento.Después de un ciclo de tratamiento de 28 días, en el momento de la concentración máxima de sorafenib, el QTcB se prolongó en 4 ± 19 mseg y el QTcF en 9 ± 18 mseg, en comparación con el valor inicial para el grupo de placebo. Ningún paciente mostró un valor de QTcB o QTcF> 500 mseg durante la monitorización del ECG posterior al tratamiento (ver sección 4.4).
Población pediátrica
La Agencia Europea de Medicamentos ha eximido de la obligación de presentar los resultados de los estudios en todos los subgrupos de la población pediátrica para el cáncer de riñón y pelvis renal (excluyendo nefroblastoma, nefroblastomatosis, sarcoma de células claras, nefroma mesoblástico, carcinoma medular renal y tumor rabdoide de riñón). y carcinoma de hígado y de vías biliares intrahepáticas (excluyendo hepatoblastoma) y carcinoma diferenciado de tiroides (ver sección 4.2 para obtener información sobre uso pediátrico).
05.2 "Propiedades farmacocinéticas
Absorción y distribución
Después de la administración de comprimidos de sorafenib, la biodisponibilidad relativa media es de 38 a 49% en comparación con una solución oral. Se desconoce la biodisponibilidad absoluta. Después de la administración oral, sorafenib alcanza los niveles plasmáticos máximos en aproximadamente 3 horas. Cuando se administra con una comida rica en grasas, la absorción de sorafenib se reduce en aproximadamente un 30% en comparación con la administración en ayunas.
La Cmax y el AUC medias aumentan menos que proporcionalmente con dosis superiores a 400 mg dos veces al día. Unión a proteínas plasmáticas de sorafenib in vitro es del 99,5%.
La dosificación repetida de sorafenib durante 7 días resultó en una acumulación de 2,5 a 7 veces en comparación con la administración única. El estado de equilibrio de sorafenib se alcanza en 7 días, con una relación entre las concentraciones plasmáticas media máxima y mínima de menos de 2.
Se determinaron las concentraciones de equilibrio de sorafenib administrado a 400 mg dos veces al día en pacientes con DTC, RCC y HCC. La concentración media más alta se observó en pacientes con DTC (aproximadamente el doble de la observada en pacientes con RCC y HCC), pero la variabilidad fue alta para Todos los tipos de tumores Se desconoce la causa de esta mayor concentración en pacientes con CDT.
Biotransformación y eliminación
La vida media de eliminación de sorafenib es de aproximadamente 25 a 48 horas. El sorafenib se metaboliza principalmente en el hígado a través del metabolismo oxidativo mediado por CYP3A4 y la conjugación de glucurono-conjugación mediada por UGT1A9. El sorafenib conjugado puede liberarse en el tracto gastrointestinal por la actividad glucuronidasa de algunas bacterias, permitiendo así la reabsorción del principio activo no conjugado.Se ha observado que la combinación con neomicina interfiere con este proceso, disminuyendo la biodisponibilidad media de sorafenib en un 54%.
El sorafenib representa aproximadamente el 70-85% de los analitos que circulan en el plasma en estado estacionario. Se han identificado ocho metabolitos de sorafenib, cinco de los cuales se han encontrado en plasma. El principal metabolito de sorafenib que circula en plasma, el N-óxido de piridina, exhibe potenciain vitro similar al de sorafenib. Este metabolito representa aproximadamente del 9 al 16% de los analitos que circulan en estado estacionario.
Tras la administración oral de una dosis de 100 mg de solución de sorafenib, se recuperó el 96% de la dosis en 14 días: 77% en heces y 19% en orina como metabolitos de glucuronato. El sorafenib inalterado, que representa el 51% de la dosis, se recuperó en las heces pero no en la orina, lo que indica que la excreción biliar del principio activo no metabolizado puede contribuir a la eliminación del sorafenib.
Farmacocinética en categorías particulares de pacientes
El análisis de los datos demográficos mostró que no existe correlación entre la farmacocinética y la edad (hasta 65 años), sexo o peso corporal.
Población pediátrica
No se han realizado estudios para verificar la farmacocinética de sorafenib en pacientes pediátricos.
Raza
No existen diferencias clínicamente relevantes en la farmacocinética entre sujetos caucásicos y asiáticos.
Insuficiencia renal
En cuatro estudios clínicos de fase I, la exposición a sorafenib en estado estacionario en pacientes con insuficiencia renal leve o moderada fue similar a la encontrada en pacientes con función renal normal. En un estudio de farmacología clínica (dosis única de 400 mg de sorafenib) no se observó relación entre exposición a sorafenib y función renal en sujetos con función renal normal o insuficiencia renal leve, moderada o grave. No hay datos disponibles en pacientes que requieran diálisis.
Deterioro hepático
En pacientes con carcinoma hepatocelular (HCC) y con la insuficiencia hepática evaluada como Child-Pugh A o B (leve a moderada), los valores de exposición fueron comparables y dentro del rango observado en pacientes sin insuficiencia hepática. La farmacocinética de sorafenib en pacientes Child-Pugh A y B sin HCC fue similar a la observada en voluntarios sanos. No hay datos para pacientes con insuficiencia hepática grave (Child-Pugh C). Sorafenib se elimina principalmente a través del hígado y la exposición puede aumentar en esta población de pacientes.
05.3 Datos preclínicos sobre seguridad
Se evaluó el perfil de seguridad preclínico de sorafenib en ratones, ratas, perros y conejos.
Los estudios de toxicidad a dosis repetidas han revelado cambios en varios órganos (degeneración y regeneración) a exposiciones por debajo de las dosis utilizadas en los estudios clínicos (basados en una comparación del AUC).
Después de la administración repetida en perros jóvenes y en crecimiento, se observaron efectos sobre los huesos y los dientes a exposiciones inferiores a las dosis utilizadas en los estudios clínicos. Estos efectos consistieron en un engrosamiento desigual de la placa de crecimiento del fémur, hipoplasia medular en la vecindad de las placas de crecimiento alteradas y cambios en la composición de la dentina. No se indujeron efectos similares en el perro adulto.
Se llevó a cabo el programa estándar de estudios de genotoxicidad y se obtuvieron resultados positivos ya que se observó un aumento en las aberraciones estructurales cromosómicas en un ensayo. in vitro en células de mamíferos (ovarios de hámster chino) para medir la clastogenicidad en presencia de activación metabólica. Sorafenib no fue genotóxico en la prueba de Ames ni en la prueba de micronúcleos en vivo en el ratón. Un intermedio del proceso de fabricación, que también está presente en el principio activo final (in vitro en células bacterianas (prueba de Ames). Además, el lote de sorafenib ensayado en la batería genotóxica estándar incluía PAPE al 0,34%.
No se han realizado estudios de carcinogenicidad con sorafenib.
No se han realizado estudios específicos en animales con sorafenib para evaluar el efecto sobre la fertilidad. Sin embargo, es de esperar un efecto adverso sobre la fertilidad masculina y femenina, ya que los estudios en animales con dosis repetidas han mostrado cambios en los órganos reproductores masculinos y femeninos a exposiciones inferiores a las dosis utilizadas en los ensayos clínicos (basadas en el AUC). signos de degeneración y retraso en el desarrollo de los testículos, epidídimo, próstata y vesículas seminales en ratas.Las ratas hembras mostraron necrosis central del cuerpo lúteo y bloqueo del desarrollo folicular en los ovarios. Los perros mostraron degeneración tubular en los ovarios, testículos y oligospermia.
Se ha demostrado que sorafenib es embriotóxico y teratogénico cuando se administra a ratas y conejos a exposiciones inferiores a las dosis utilizadas en los estudios clínicos. Los efectos observados incluyeron una disminución en el peso corporal materno y fetal, un aumento en el número de reabsorciones fetales y un mayor número de malformaciones externas y viscerales.
Los estudios de evaluación del riesgo ambiental han demostrado que el tosilato de sorafenib es potencialmente persistente, bioacumulativo y tóxico para el medio ambiente. La información sobre la evaluación del riesgo ambiental está disponible en el Informe público europeo de evaluación (EPAR) de este medicamento (ver sección 6.6).
06.0 INFORMACIÓN FARMACÉUTICA
06.1 Excipientes
Núcleo de la tableta:
Croscarmelosa sódica
Celulosa microcristalina
Hipromelosa
Lauril Sulfato de Sodio
Estearato de magnesio
Recubrimiento de tabletas:
Hipromelosa
Macrogol
Dióxido de titanio (E 171)
Óxido de hierro rojo (E 172)
06.2 Incompatibilidad
Irrelevante.
06.3 Período de validez
3 años.
06.4 Precauciones especiales de conservación
No conservar por encima de 25 ° C.
06.5 Naturaleza del envase primario y contenido del envase.
Caja que contiene 112 comprimidos recubiertos con película (4 x 28) en blíster transparente (PP / Aluminio).
06.6 Instrucciones de uso y manipulación
Este medicamento puede suponer un riesgo potencial para el medio ambiente El medicamento no utilizado y los desechos derivados de este medicamento deben eliminarse de acuerdo con las normativas locales.
07.0 TITULAR DE LA AUTORIZACIÓN DE COMERCIALIZACIÓN
Bayer Pharma AG
13342 Berlín
Alemania
08.0 NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN
EU / 1/06/342/001
037154010
09.0 FECHA DE LA PRIMERA AUTORIZACIÓN O RENOVACIÓN DE LA AUTORIZACIÓN
Fecha de la primera autorización: 19 de julio de 2006
Fecha de la última renovación: 21 de julio de 2011
10.0 FECHA DE REVISIÓN DEL TEXTO
05/2014