Ingredientes activos: Ustekinumab
STELARA 45 mg solución inyectable
Los prospectos de Stelara están disponibles para los siguientes tamaños de envase:- STELARA 45 mg solución inyectable
- STELARA 90 mg solución inyectable
Indicaciones ¿Por qué se usa Stelara? ¿Para qué sirve?
Que es Stelara
Stelara contiene el principio activo "ustekinumab", un anticuerpo monoclonal.
Los anticuerpos monoclonales son proteínas que reconocen y se unen a determinadas proteínas del organismo. Stelara pertenece a un grupo de medicamentos denominados "inmunosupresores". Estos medicamentos reducen la actividad del sistema inmunológico hasta cierto punto.
Para que sirve Stelara
Stelara se utiliza para tratar las siguientes enfermedades inflamatorias:
- psoriasis en placas (en adultos y niños a partir de los 12 años)
- artritis psoriásica (en adultos)
Soriasis en placas
La psoriasis en placas es una afección cutánea que causa inflamación de la piel y las uñas. Stelara reducirá la inflamación y otros signos de la enfermedad.
Stelara se usa en adultos con psoriasis en placas de moderada a grave, que no pueden usar ciclosporina, metotrexato o fototerapia, o para quienes estos tratamientos no funcionan.
Stelara se utiliza en niños a partir de los 12 años con psoriasis en placas de moderada a grave que no pueden tolerar la fototerapia u otras terapias sistémicas, o cuando estos tratamientos no han funcionado.
Artritis psoriásica
La artritis psoriásica es una enfermedad inflamatoria de las articulaciones, generalmente acompañada de psoriasis. Si tiene artritis psoriásica activa, primero se le tratará con otros medicamentos. Si no responde adecuadamente a estos medicamentos, puede tomar Stelara para:
- reducir los signos y síntomas de la enfermedad.
- mejorar la función física.
- ralentizar el daño a las articulaciones.
Contraindicaciones Cuándo no se debe usar Stelara
No use Stelara
- Si es alérgico a ustekinumab oa cualquiera de los demás componentes de este medicamento (incluidos en la sección 6 a continuación).
- Si tiene una infección activa que su médico considere importante.
Si no está seguro de si algo de lo anterior le aplica, hable con su médico o farmacéutico antes de usar Stelara.
Precauciones de uso Lo que necesita saber antes de tomar Stelara
Hable con su médico o farmacéutico antes de usar Stelara. Su médico controlará su salud antes de cada tratamiento. Asegúrese de informar a su médico antes de cualquier tratamiento sobre las enfermedades que padece. Además, informe a su médico incluso si recientemente ha estado en contacto con personas que pueden haber tenido tuberculosis. Su médico lo examinará y realizará pruebas de tuberculosis antes de administrarle Stelara. Si su médico cree que tiene riesgo de contraer tuberculosis, es posible que le recete medicamentos para tratar la tuberculosis.
Tenga cuidado con los efectos secundarios graves
Stelara puede provocar efectos secundarios graves, incluidas reacciones alérgicas e infecciones. Debe prestar atención a ciertos signos de la enfermedad mientras toma Stelara. Consulte "Efectos secundarios graves" en la sección 4 para obtener una lista completa de estos efectos secundarios.
Antes de usar Stelara, comuníquese con su médico:
- Si alguna vez ha tenido una reacción alérgica a Stelara. Pregúntele a su médico si no está seguro.
- Si alguna vez ha tenido algún tipo de cáncer, esto se debe a que los inmunosupresores como Stelara debilitan parcialmente el sistema inmunológico. Esto puede aumentar el riesgo de cáncer.
- Si tiene o ha tenido una infección reciente.
- Si alguna vez ha tenido lesiones nuevas o que han cambiado en el área de la psoriasis o en la piel normal.
- Si está tomando cualquier otro tipo de tratamiento para la psoriasis y / o la artritis psoriásica, como otro inmunosupresor o fototerapia (cuando el cuerpo se trata con un tipo de luz ultravioleta (UV)). Estos tratamientos también pueden reducir en parte la actividad del sistema inmunológico No se ha estudiado el uso concomitante de estas terapias con Stelara. Sin embargo, es posible que aumente la posibilidad de enfermedades relacionadas con un debilitamiento del sistema inmunológico.
- Si está usando o ha usado alguna vez inyecciones para tratar alergias, no se sabe si Stelara puede afectarlas.
- Si tiene 65 años o más, es más probable que tenga infecciones.
Si no está seguro de si se encuentra en alguna de las condiciones anteriores, consulte a su médico o farmacéutico antes de recibir tratamiento con Stelara.
Niños y adolescentes
Stelara no se recomienda para el tratamiento de niños (menores de 12 años) porque no se ha estudiado en este grupo de edad.
Interacciones ¿Qué medicamentos o alimentos pueden cambiar el efecto de Stelara?
Informe a su médico o farmacéutico:
- Si está tomando, ha tomado recientemente o podría tomar cualquier otro medicamento.
- Si ha sido vacunado recientemente o está a punto de vacunarse. Algunos tipos de vacunas (vacunas vivas) no deben administrarse mientras esté usando Stelara.
Advertencias Es importante saber que:
Embarazo y lactancia
- Es preferible evitar el uso de Stelara durante el embarazo. Se desconocen los efectos de Stelara en mujeres embarazadas. Si está en edad fértil, es aconsejable evitar quedarse embarazada; debe usar un método anticonceptivo adecuado mientras usa Stelara y durante al menos 15 semanas después de interrumpir el tratamiento con Stelara. Si está embarazada, cree que podría estarlo o tiene intención de quedarse embarazada, consulte a su médico.
- Si está amamantando o planea hacerlo, consulte a su médico. Usted y su médico decidirán si debe amamantar o usar Stelara. No puede hacer ambas cosas.
Conducción y uso de máquinas
La influencia de Stelara sobre la capacidad para conducir o utilizar máquinas es nula o insignificante.
Dosis, método y momento de administración Cómo usar Stelara: Posología
Stelara está diseñado para su uso bajo la guía y supervisión de un médico con experiencia en el diagnóstico y tratamiento de la psoriasis o la artritis psoriásica. Siga exactamente las instrucciones de administración de este medicamento indicadas por su médico. En caso de duda, consulte a su médico. Hable con su médico sobre cuándo necesitará las inyecciones y las visitas de seguimiento.
Cuánto Stelara se administra
Su médico decidirá cuánto Stelara necesita y durante cuánto tiempo.
Adultos a partir de 18 años
- La dosis inicial recomendada es de 45 mg de Stelara. Los pacientes que pesen más de 100 kilogramos (kg) pueden comenzar con una dosis de 90 mg en lugar de 45 mg.
- Después de la dosis inicial, tomará la siguiente dosis 4 semanas después y luego cada 12 semanas. Las dosis posteriores suelen ser las mismas que la dosis inicial.
Niños y adolescentes a partir de 12 años
- Su médico calculará la dosis correcta, incluida la cantidad (volumen) de Stelara que debe inyectarse para asegurarse de que se administra la dosis correcta. La dosis correcta dependerá del peso corporal del niño en el momento de cada dosis.
- Si su peso corporal es inferior a 60 kg, la dosis recomendada es de 0,75 mg de Stelara por kg de peso corporal.
- Si el peso corporal se encuentra entre 60 kg y 100 kg, la dosis recomendada es de 45 mg de Stelara.
- Si el peso supera los 100 kg, la dosis recomendada es de 90 mg de Stelara.
- Después de la dosis inicial, deberá recibir la siguiente dosis después de 4 semanas y, posteriormente, cada 12 semanas.
Cómo se administra Stelara
- Stelara se administra como una 'inyección debajo de la piel (' por vía subcutánea '). Al comienzo del tratamiento, su médico o enfermero puede inyectar Stelara.
- Sin embargo, usted y su médico pueden decidir si puede inyectarse Stelara usted mismo. En este caso, se le enseñará a inyectarse Stelara usted mismo.
- Para obtener instrucciones sobre cómo inyectar Stelara, consulte las "Instrucciones de administración" al final de este prospecto.
Informe a su médico si tiene alguna pregunta sobre cómo inyectarse usted mismo.
Si olvidó usar Stelara
Si olvida una dosis, comuníquese con su médico o farmacéutico. No tome una dosis doble para compensar las dosis olvidadas.
Si deja de tomar Stelara
No es peligroso dejar de usar Stelara, sin embargo, si interrumpe el tratamiento, su psoriasis puede reaparecer.
Si tiene más preguntas sobre el uso de este medicamento, consulte a su médico o farmacéutico.
Sobredosis Qué hacer si ha tomado demasiado Stelara
Si ha usado o recibido demasiado Stelara, informe a su médico o farmacéutico de inmediato. Lleve siempre consigo el embalaje exterior de su medicamento, incluso si está vacío.
Efectos secundarios ¿Cuáles son los efectos secundarios de Stelara?
Como todos los medicamentos, este medicamento puede producir efectos adversos, aunque no todas las personas los sufran.
Efectos secundarios graves
Algunos pacientes pueden experimentar efectos secundarios graves que pueden requerir tratamiento urgente.
Reacciones alérgicas: pueden necesitar tratamiento urgente, por lo tanto, comuníquese con su médico o busque ayuda médica de emergencia si nota alguno de los siguientes signos.
- Las reacciones alérgicas graves ("anafilaxia") son raras en pacientes que toman Stelara (afectan hasta 1 de cada 1.000 pacientes). Los signos incluyen:
- dificultad para respirar o tragar
- presión arterial baja, que puede causar mareos
- sensación de mareo o hinchazón en la cara, labios, boca o garganta.
- Los signos comunes de una reacción alérgica incluyen erupciones cutáneas y urticaria (que afectan hasta 1 de cada 100 personas).
Si tiene una reacción alérgica grave, su médico puede decidir que no debe volver a utilizar Stelara.
Infecciones: es posible que necesiten un tratamiento urgente, por lo tanto, comuníquese con su médico de inmediato si nota alguno de los siguientes signos.
- Las infecciones de nariz y garganta y el resfriado común son comunes (afectan hasta 1 de cada 10 personas).
- La "inflamación del tejido subcutáneo ('celulitis") es poco común (afecta hasta 1 de cada 100 pacientes).
- El herpes zooster (un tipo de erupción con ampollas) es poco común (afecta hasta 1 de cada 100 pacientes).
Stelara puede disminuir la capacidad de combatir infecciones y algunas infecciones pueden volverse graves.
Debe prestar atención a los signos de infección mientras usa Stelara. Éstos incluyen:
- fiebre, síntomas similares a los de la gripe, sudores nocturnos
- sensación de cansancio o falta de aire, tos persistente
- piel caliente, enrojecida, dolorida o una erupción dolorosa y con ampollas
- ardor al orinar
- Diarrea
Informe a su médico de inmediato si nota alguno de estos signos de infección. Hable con su médico si tiene algún tipo de infección que persiste o vuelve a aparecer. Su médico puede decidir suspender Stelara hasta que la infección desaparezca. También informe a su médico si tiene cortes abiertos o heridas que puedan infectarse.
Exfoliación de la piel: el aumento del enrojecimiento y la descamación de la piel en una gran área del cuerpo pueden ser síntomas de psoriasis eritrodérmica o dermatitis exfoliativa, que son afecciones graves de la piel. Si nota alguno de estos signos, debe informar a su médico de inmediato.
Otros efectos secundarios
Efectos adversos frecuentes (afectan hasta 1 de cada 10 pacientes):
- Diarrea
- Náusea
- Sensación de cansancio
- Sintiéndose mareado
- Dolor de cabeza
- Picor
- Dolor de espalda, músculos o articulaciones.
- Dolor de garganta
- Infección dental
- Enrojecimiento y dolor en el lugar de la inyección.
Efectos adversos poco frecuentes (afectan hasta 1 de cada 100 pacientes):
- Depresión
- Nariz congestionada o que moquea
- Sangrado, hematomas, rigidez, hinchazón y picazón en el lugar de la inyección.
- Párpado caído y relajación de los músculos de un lado de la cara ("parálisis facial" o "parálisis de Bell"), que suele ser temporal
- Un cambio en la psoriasis con enrojecimiento y nuevas ampollas en la piel pequeñas, amarillas o blancas, a veces acompañadas de fiebre (psoriasis pustulosa).
- Descamación de la piel (exfoliación de la piel)
Efectos adversos raros (afectan hasta 1 de cada 1.000 pacientes)
- Enrojecimiento y descamación de la piel sobre una gran superficie del cuerpo, que puede causar picazón o dolor (dermatitis exfoliativa). A veces se desarrollan síntomas similares como una progresión natural en el tipo de síntomas de la psoriasis (psoriasis eritrodérmica).
Notificación de efectos secundarios
Si experimenta cualquier efecto adverso, consulte a su médico o farmacéutico, incluido cualquier posible efecto adverso no mencionado en este prospecto. También puede notificar los efectos secundarios directamente a través del sistema de notificación nacional que figura en el Apéndice V. Al notificar los efectos secundarios, puede ayudar a proporcionar más información sobre la seguridad de este medicamento.
Caducidad y retención
- Mantenga este medicamento fuera de la vista y del alcance de los niños.
- Conservar en nevera (entre 2 ° C y 8 ° C). No congelar.
- Mantenga el vial en el embalaje exterior para proteger el medicamento de la luz.
- No agite los viales de Stelara. La agitación vigorosa prolongada puede dañar el medicamento.
No use este medicamento
- Después de la fecha de caducidad que se indica en la etiqueta y la caja después de "CAD" La fecha de caducidad se refiere al último día del mes.
- Si el líquido está descolorido, es opaco o si observa partículas extrañas flotantes (ver sección 6 "Descripción de cómo es Stelara y contenido del envase").
- Si sabe o cree que el medicamento ha estado expuesto a temperaturas extremas (por ejemplo, se ha congelado o calentado accidentalmente).
- Si el producto se ha agitado vigorosamente.
- Si el sello está roto.
Stelara es para un solo uso. Cualquier producto no utilizado que quede en el vial y la jeringa debe desecharse.
Los medicamentos no se deben tirar por los desagües ni a la basura. Pregunte a su farmacéutico cómo deshacerse de los envases y de los medicamentos que no necesita, ya que esto ayudará a proteger el medio ambiente.
Fecha límite "> Otra información
Qué contiene Stelara
- El ingrediente activo es ustekinumab. Cada vial contiene 45 mg de ustekinumab en 0,5 ml.
- Los demás componentes son: L-histidina, monohidrocloruro de L-histidina monohidrato, polisorbato 80, sacarosa, agua para preparaciones inyectables.
Aspecto de Stelara y contenido del envase
Stelara es una solución inyectable de transparente a ligeramente opalescente (apariencia perlada), de incolora a amarillo pálido.
La solución puede contener algunas pequeñas partículas de proteína translúcidas o blancas. Se presenta en un envase de cartón que contiene 1 dosis única, en un vial de vidrio de 2 ml.
Cada vial contiene 45 mg de ustekinumab en 0,5 ml de solución inyectable.
Caducidad "> Instrucciones de administración
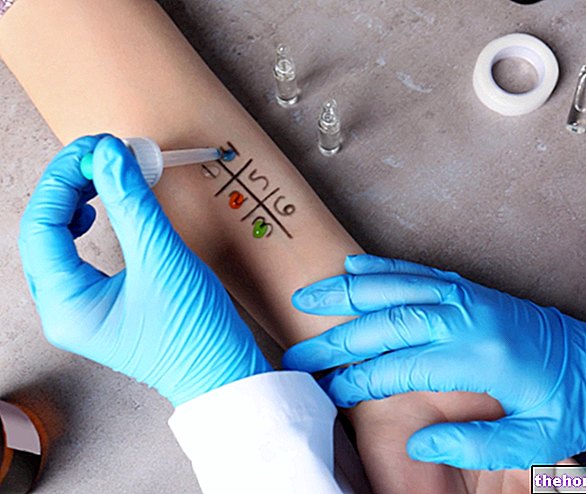
Al comienzo del tratamiento, su médico le ayudará durante la primera inyección. Sin embargo, usted y su médico pueden decidir si puede inyectarse Stelara usted mismo. En este caso, se le enseñará cómo inyectarse Stelara usted mismo. Informe a su médico en caso de si tiene alguna pregunta sobre cómo inyectarse usted mismo.
- No mezcle Stelara con otros líquidos inyectables.
- No agite los viales de Stelara, ya que agitarlos vigorosamente puede dañar el medicamento. No use el medicamento si se ha agitado vigorosamente.
Verifique el número de viales y prepare los materiales:
Saque el vial o varios viales del frigorífico. Deje el vial fuera del refrigerador durante aproximadamente media hora, esto permitirá que el líquido alcance una temperatura agradable para la inyección (temperatura ambiente).
Mira esto:
- el número de viales y la dosis son correctos
- si su dosis es de 45 mg o menos, tomará un vial de 45 mg de Stelara
- si su dosis es de 90 mg, tomará dos viales de 45 mg de Stelara y deberá administrar dos inyecciones. Elija dos lugares diferentes del cuerpo para estas inyecciones (por ejemplo, una inyección en el muslo derecho y la otra inyección en el muslo izquierdo) y proceda con las inyecciones una tras otra. Utilice una nueva aguja y una nueva jeringa para cada inyección.
- la medicina es correcta
- el medicamento no ha caducado
- el vial no está dañado y el tapón está roto
- la solución en el vial es transparente o ligeramente opalescente (apariencia perlada) e incolora o de color amarillo pálido
- el líquido no tiene un color alterado u opaco y no contiene partículas extrañas
- no está congelado.
Los niños con un peso corporal de menos de 60 kg necesitan una dosis de menos de 45 mg. Debe estar seguro de la cantidad (volumen) adecuada que debe extraer del vial y del tipo de jeringa necesaria para la dosificación. Si no sabe la cantidad de medicamento o el tipo de jeringa que debe usar, comuníquese con su médico para obtener más instrucciones.
Tome todo lo que necesite y colóquelo sobre una superficie limpia. Debe haber una jeringa, aguja, hisopos antisépticos, una bola de algodón o gasa y un recipiente para objetos punzantes.
Elija el lugar de la inyección y prepárelo:
Elija un lugar de inyección.
- Stelara se administra mediante inyección debajo de la piel (vía subcutánea).
- Un buen lugar para la inyección es la parte superior del muslo o alrededor del vientre (abdomen) al menos a 5 cm del ombligo.
- Si es posible, no elija áreas de la piel con signos de psoriasis.
- Si alguien lo está ayudando durante la inyección, también puede elegir la parte superior de los brazos como lugar de inyección.
Prepare el lugar de la inyección
- Lávate muy bien las manos con jabón y agua tibia.
- Frote el lugar de la inyección en la piel con un hisopo antiséptico.
- No vuelva a tocar esta zona antes de inyectarse.
Prepare la dosis:
- Retire la tapa de la parte superior del vial.
- No quite la tapa
- Limpiar la tapa con un hisopo antiséptico.
- Coloque el vial sobre una superficie plana.
- Tome la jeringa y retire el capuchón protector de la aguja.
- No toque la aguja ni deje que la aguja toque nada.
- Empuje la aguja a través del tapón de goma.
- Ponga el vial y la jeringa boca abajo.
- Tire del émbolo de la jeringa para llenar la jeringa con la cantidad de líquido recetada por su médico.
- Es importante que la aguja esté siempre dentro del líquido para que no se formen burbujas de aire en la jeringa.
- Retire la aguja del vial.
- Sostenga la jeringa con la aguja apuntando hacia arriba para ver si hay burbujas en el interior.
- Si hay burbujas de aire, golpee suavemente el costado de la jeringa hasta que las burbujas de aire lleguen a la parte superior de la jeringa.
- A continuación, presione el émbolo hasta que se haya eliminado todo el aire (pero no el líquido), no deje reposar la jeringa y evite que la aguja toque nada.
Inyecte la dosis:
- Apriete suavemente la parte de piel limpia sosteniéndola entre el pulgar y el índice, sin apretar demasiado.
- Introduzca la aguja en la piel pellizcada.
- Empuje el émbolo con el pulgar hasta que termine de inyectar todo el líquido. Presione lenta y constantemente, manteniendo la piel suavemente tensa.
- Cuando el émbolo llegue al final de la jeringa, saque la aguja y suelte la piel.
Después de la inyección:
- Presione una almohadilla antiséptica sobre el lugar de la inyección durante unos segundos después de la inyección.
- Puede haber una pequeña cantidad de sangre o líquido en el lugar de la inyección. Es normal.
- Puede presionar una bola de algodón o una gasa en el lugar de la inyección y mantenerla durante 10 segundos.
- No frote la piel en el lugar de la inyección; puede cubrir el lugar de la inyección con un parche pequeño si es necesario.
Disposición:
- Las jeringas y agujas usadas deben colocarse en un recipiente resistente a perforaciones, como un recipiente para objetos punzantes. Por su salud y seguridad y la seguridad de los demás, nunca reutilice agujas o jeringas. Deseche el recipiente para objetos punzantes de acuerdo con las normativas locales.
- Los viales vacíos, las toallitas antisépticas y otros dispositivos pueden desecharse en la basura.
Prospecto fuente: AIFA (Agencia Italiana de Medicamentos). Contenido publicado en enero de 2016. Es posible que la información presente no esté actualizada.
Para tener acceso a la versión más actualizada, es recomendable acceder al sitio web de la AIFA (Agencia Italiana de Medicamentos). Descargo de responsabilidad e información útil.
01.0 NOMBRE DEL MEDICAMENTO -
SOLUCIÓN STELARA PARA INYECCIÓN
02.0 COMPOSICIÓN CUALITATIVA Y CUANTITATIVA -
STELARA 45 mg solución inyectable
Cada vial contiene 45 mg de ustekinumab en 0,5 ml.
STELARA 90 mg solución inyectable
Cada vial contiene 90 mg de ustekinumab en 1 ml.
STELARA 45 mg solución inyectable en jeringa precargada
Cada jeringa precargada contiene 45 mg de ustekinumab en 0,5 ml.
STELARA 90 mg solución inyectable en jeringa precargada
Cada jeringa precargada contiene 90 mg de ustekinumab en 1 ml.
Ustekinumab es un anticuerpo monoclonal IgG1κ de unión a interleucina (IL) -12/23 totalmente humano producido en una línea celular de mieloma de ratón utilizando tecnología de ADN recombinante.
Para consultar la lista completa de excipientes, ver sección 6.1.
03.0 FORMA FARMACÉUTICA -
STELARA 45 mg solución inyectable
Solución inyectable.
STELARA 90 mg solución inyectable
Solución inyectable.
STELARA 45 mg solución inyectable en jeringa precargada
Solución inyectable.
STELARA 90 mg solución inyectable en jeringa precargada
Solución inyectable.
La solución es de transparente a ligeramente opalescente, de incolora a amarillo pálido.
04.0 INFORMACIÓN CLÍNICA -
04.1 Indicaciones terapéuticas -
Soriasis en placas
STELARA está indicado para el tratamiento de la psoriasis en placas de moderada a grave en pacientes adultos que no han respondido, que tienen contraindicaciones o que son intolerantes a otras terapias sistémicas, como ciclosporina, metotrexato (MTX) o PUVA (psoraleno y rayos ultravioleta A) ( ver sección 5.1).
Psoriasis en placas en pacientes pediátricos
STELARA está indicado para el tratamiento de la psoriasis en placas de moderada a grave en pacientes adolescentes a partir de los 12 años que no están adecuadamente controlados o son intolerantes a otras terapias sistémicas o fototerapia (ver sección 5.1).
Artritis psoriásica (PsA)
STELARA, solo o en combinación con MTX, está indicado para el tratamiento de la artritis psoriásica activa en pacientes adultos cuando la respuesta al tratamiento previo con fármacos antirreumáticos modificadores de la enfermedad (FAME) no biológicos ha sido inadecuada (ver sección 5.1).
enfermedad de Crohn
STELARA está indicado para el tratamiento de pacientes adultos con enfermedad de Crohn activa de moderada a grave que han tenido una respuesta inadecuada, han perdido la respuesta o se han encontrado intolerantes a la terapia convencional o un antagonista del TNFα o que tienen contraindicaciones para dichas terapias.
04.2 Posología y forma de administración -
STELARA debe utilizarse bajo la guía y supervisión de médicos especialistas con experiencia en el diagnóstico y tratamiento de las afecciones para las que está indicado STELARA.
Dosis
Soriasis en placas
La posología recomendada de STELARA es una dosis inicial de 45 mg administrada por vía subcutánea, seguida de una dosis de 45 mg después de 4 semanas y cada 12 semanas a partir de entonces.
Se debe considerar la interrupción del tratamiento en pacientes que no han mostrado respuesta a las 28 semanas de tratamiento.
Pacientes con peso corporal> 100 kg
En pacientes que pesen más de 100 kg, la dosis inicial que se administrará por vía subcutánea es de 90 mg, seguida de una dosis de 90 mg después de 4 semanas y cada 12 semanas a partir de entonces. También se ha demostrado que la dosis de 45 mg es eficaz en estos pacientes. Sin embargo, la dosis de 90 mg mostró una mayor eficacia (ver sección 5.1, Tabla 4).
Artritis psoriásica (PsA)
La posología recomendada de STELARA es una dosis inicial de 45 mg administrada por vía subcutánea, seguida de una dosis de 45 mg después de 4 semanas y cada 12 semanas a partir de entonces. Alternativamente, se pueden usar 90 mg en pacientes con un peso corporal> 100 kg. Se debe considerar la interrupción del tratamiento en pacientes que no han mostrado respuesta a las 28 semanas de tratamiento.
Ancianos (≥ 65 años)
No es necesario un ajuste de dosis en pacientes de edad avanzada (ver sección 4.4).
Insuficiencia renal y hepática
STELARA no se ha estudiado en esta población de pacientes. No se puede hacer ninguna recomendación sobre la dosis a administrar.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de STELARA en niños con psoriasis menores de 12 años o en niños con artritis psoriásica menores de 18 años.
Psoriasis en placas en pacientes pediátricos (a partir de los 12 años)
La dosis recomendada de STELARA basada en el peso corporal se muestra en las tablas siguientes (Tablas 1 y 2). STELARA debe administrarse en las semanas 0 y 4 y, a partir de entonces, cada 12 semanas.
Tabla 1: Dosis recomendada de STELARA para pacientes pediátricos con psoriasis
a Para calcular el volumen de inyección (ml) para el peso corporal del paciente (kg) x 0,0083 (ml / kg) o consulte la Tabla 2. El volumen calculado debe redondearse al 0,01 ml más cercano y administrarse con una jeringa graduada de 1 ml. Hay un vial de 45 mg disponible para pacientes pediátricos que necesitan recibir menos de la dosis completa de 45 mg. .
Tabla 2: Volúmenes de inyección de STELARA para pacientes pediátricos
Se debe considerar la interrupción del tratamiento en pacientes que no muestran una respuesta hasta las 28 semanas de tratamiento.
enfermedad de Crohn
En el régimen de tratamiento, la primera dosis de STELARA se administra por vía intravenosa. Para conocer la posología de la pauta posológica intravenosa, ver la sección 4.2 de la ficha técnica de STELARA 130 mg concentrado para solución para perfusión.
La primera administración subcutánea de 90 mg de STELARA debe ocurrir en la semana 8 después de la dosis intravenosa. Después de esto, se recomienda la dosificación cada 12 semanas.
Los pacientes que no hayan mostrado una respuesta adecuada 8 semanas después de la primera dosis subcutánea pueden recibir una segunda dosis subcutánea (ver sección 5.1).
Los pacientes que no han administrado la dosis cada 12 semanas pueden beneficiarse de un aumento de la frecuencia de administración cada 8 semanas (ver sección 5.1).
Los pacientes pueden recibir la dosis cada 8 semanas o cada 12 semanas a partir de entonces en función del criterio clínico (ver sección 5.1).
Se debe considerar la interrupción del tratamiento en pacientes que no muestran evidencia de beneficio terapéutico en la semana 16 o en la semana 16 después de cambiar a la dosis de cada 8 semanas.
Los inmunomoduladores y / o corticosteroides pueden continuarse durante el tratamiento con STELARA. En pacientes que han respondido al tratamiento con STELARA, los corticosteroides pueden reducirse o retirarse de acuerdo con el tratamiento estándar.
Si se interrumpe el tratamiento, es seguro y eficaz reanudar el tratamiento con la administración subcutánea cada 8 semanas.
Ancianos (≥ 65 años)
No es necesario un ajuste de dosis en pacientes de edad avanzada (ver sección 4.4).
Insuficiencia renal y hepática
STELARA no se ha estudiado en esta población de pacientes. No se puede hacer ninguna recomendación sobre la dosis a administrar.
Población pediátrica
No se ha establecido todavía la seguridad y eficacia de STELARA en el tratamiento de la enfermedad de Crohn en niños menores de 18 años No hay datos disponibles.
Método de administración
STELARA 45 mg y 90 mg en viales o jeringas precargadas está formulado solo para inyección subcutánea. Si es posible, evite la inyección en áreas afectadas por psoriasis.
Después de recibir las instrucciones adecuadas sobre la técnica de inyección subcutánea, los pacientes o sus cuidadores pueden administrar STELARA si el médico lo considera apropiado. Sin embargo, el médico debe garantizar un seguimiento periódico adecuado de los pacientes. Se debe indicar a los pacientes o cuidadores que administren la cantidad prescrita de STELARA como se indica en el prospecto. Las instrucciones completas de administración se proporcionan en el prospecto.
Para obtener más información sobre la preparación y las precauciones especiales de manipulación, ver sección 6.6.
04.3 Contraindicaciones -
Hipersensibilidad al principio activo oa alguno de los excipientes incluidos en la sección 6.1.
Infección activa clínicamente relevante (por ejemplo, tuberculosis activa; ver sección 4.4).
04.4 Advertencias especiales y precauciones de uso apropiadas -
Infecciones
Ustekinumab puede aumentar el riesgo de contraer infecciones y reactivar las latentes.
En algunos estudios clínicos, se han observado infecciones bacterianas, fúngicas y víricas graves en pacientes que reciben STELARA (ver sección 4.8).
Se debe tener precaución al considerar el uso de STELARA en pacientes con una infección crónica o con antecedentes de infección recurrente (ver sección 4.3).
Antes de iniciar el tratamiento con STELARA, se debe evaluar a todos los pacientes para detectar la presencia de infección por tuberculosis. STELARA no debe administrarse a pacientes con tuberculosis activa (ver sección 4.3). El tratamiento de la infección tuberculosa latente debe iniciarse antes de administrar STELARA. Se debe considerar la terapia antituberculosa antes de iniciar STELARA en pacientes con antecedentes de tuberculosis latente o activa que no se pueden confirmar. Se puede confirmar la vía terapéutica adecuada. Los pacientes en tratamiento con STELARA deben ser cuidadosamente monitoreado para detectar signos y síntomas de tuberculosis activa, durante y después del tratamiento.
Se debe advertir a los pacientes que busquen consejo médico si observan signos y síntomas que puedan indicar una "infección en curso. Si un paciente desarrolla una infección grave", debe ser monitoreado de cerca y STELARA no debe administrarse hasta que "la infección no desaparezca".
Neoplasias
Los inmunosupresores como ustekinumab pueden aumentar el riesgo de desarrollar cáncer.
Algunos pacientes que recibieron STELARA en ensayos clínicos desarrollaron neoplasias cutáneas y no cutáneas (ver sección 4.8).
No se han realizado estudios clínicos que incluyan pacientes con antecedentes de neoplasias malignas o en los que el tratamiento con STELARA continúe a pesar de la aparición de neoplasias malignas en curso. Por lo tanto, se debe tener precaución al considerar el tratamiento con STELARA en estos pacientes.
Todos los pacientes, especialmente los mayores de 60 años, los pacientes con antecedentes de terapia inmunosupresora prolongada o con antecedentes de tratamiento con PUVA, deben ser monitorizados para detectar cáncer de piel no melanoma (ver sección 4.8).
Reacciones hipersensibles
Se han notificado reacciones de hipersensibilidad graves en la experiencia postcomercialización, en algunos casos incluso varios días después del tratamiento. Se han producido anafilaxia y angioedema. Se debe interrumpir el tratamiento y la administración adecuados de STELARA (ver sección 4.8).
Sensibilidad al látex
La tapa de la aguja de la jeringa precargada de STELARA está hecha de caucho natural seco (un derivado del látex) que puede causar reacciones alérgicas en personas sensibles al látex.
Vacunas
Se recomienda no administrar vacunas virales o bacterianas vivas (como el bacilo de Calmette y Guérin, BCG) al mismo tiempo que el tratamiento con STELARA. No se han realizado estudios clínicos específicos en pacientes que hayan recibido recientemente vacunas virales o bacterianas vivas. No hay datos sobre la transmisión secundaria de infecciones por vacunas vivas en pacientes que reciben STELARA. Antes de administrar una vacuna vírica o bacteriana viva, el tratamiento con STELARA debe suspenderse durante al menos 15 semanas después de la última administración y no puede reanudarse antes de 2 semanas después de la vacunación. El médico prescriptor debe consultar el Resumen de las Características del Producto de la vacuna, para beneficiarse de datos adicionales y orientación sobre el uso concomitante de agentes inmunosupresores posvacunación.
Los pacientes en tratamiento con STELARA pueden tratarse simultáneamente con vacunas inactivadas o muertas.
El tratamiento a largo plazo con STELARA no suprime la respuesta inmune humoral al polisacárido neumocócico ni a la vacuna antitetánica (ver sección 5.1).
Terapia inmunosupresora concomitante
La seguridad y eficacia de STELARA en combinación con otros inmunosupresores, incluidos agentes biológicos o fototerapia, no se han evaluado en estudios de psoriasis. En estudios clínicos de artritis psoriásica, no se ha demostrado que el uso concomitante de MTX afecte la seguridad. O la eficacia de STELARA. En los estudios de la enfermedad de Crohn, el uso concomitante de inmunosupresores o corticosteroides no pareció afectar la seguridad o eficacia de STELARA.
Se debe tener precaución al considerar el uso concomitante de otros inmunosupresores y STELARA, o cuando sea resultado del tratamiento con otros inmunosupresores biológicos (ver sección 4.5).
Inmunoterapia
STELARA no se ha evaluado en pacientes que se hayan sometido a inmunoterapia para alergias.
Se desconoce si STELARA puede afectar a la inmunoterapia para alergias.
Condiciones severas de la piel.
En pacientes con psoriasis, se ha notificado dermatitis exfoliativa después del tratamiento con ustekinumab (ver sección 4.8). Los pacientes con psoriasis en placas pueden desarrollar psoriasis eritrodérmica, con síntomas que pueden ser clínicamente indistinguibles de la dermatitis exfoliativa, como un curso natural de la enfermedad. Como parte del seguimiento de los pacientes con psoriasis, los médicos deben prestar atención a los síntomas de la psoriasis eritrodérmica o la dermatitis exfoliativa. Si se presentan estos síntomas, se debe instituir la terapia adecuada. STELARA debe suspenderse si se sospecha una reacción a un medicamento.
Poblaciones especiales
Ancianos (≥ 65 años)
En general, no se observaron diferencias en la eficacia o seguridad de STELARA en pacientes de 65 años o más en comparación con pacientes más jóvenes, sin embargo, el número de pacientes de 65 años o más no es suficiente para determinar si responden de manera diferente a los pacientes más jóvenes Debido a la mayor incidencia de infecciones en la población de edad avanzada en general, se debe tener precaución al tratar a pacientes de edad avanzada.
04.5 Interacciones con otros medicamentos y otras formas de interacción -
No se deben administrar vacunas vivas al mismo tiempo que STELARA (ver sección 4.4).
No se han realizado estudios de interacción en humanos. En los análisis farmacocinéticos poblacionales de los estudios de fase III, se examinó el efecto de los medicamentos concomitantes más utilizados en pacientes con psoriasis (incluidos paracetamol, ibuprofeno, ácido acetilsalicílico)., Metformina, atorvastatina, levotiroxina ) sobre el perfil farmacocinético de ustekinumab. No se encontró interacción con estos medicamentos administrados concomitantemente. La base de este análisis fue la presencia de al menos 100 pacientes (> 5% de la población del estudio) tratados concomitantemente con estos medicamentos durante al menos el 90% del período de estudio. En pacientes con artritis psoriásica o enfermedad de Crohn, la farmacocinética de ustekinumab no se vio afectada por el uso concomitante de MTX, AINE, 6-mercaptopurina, azatioprina y corticosteroides orales, o por la exposición previa a agentes anti-TNFα. De un estudio in vitro no indican la necesidad de ajustar la dosis en pacientes que toman sustratos del CYP450 de forma concomitante (ver sección 5.2).
En estudios de psoriasis, no se han evaluado los perfiles de seguridad y eficacia de STELARA, administrado en combinación con inmunosupresores, incluidos agentes biológicos o fototerapia. En estudios de artritis psoriásica, el uso concomitante de MTX no pareció afectar la seguridad y eficacia de STELARA En los estudios de enfermedad de Crohn, el uso concomitante de inmunosupresores o corticosteroides no pareció afectar la seguridad o eficacia de STELARA (ver sección 4.4).
04.6 Embarazo y lactancia -
Mujeres en edad fértil
Las mujeres en edad fértil deben utilizar métodos anticonceptivos eficaces durante el tratamiento y durante al menos 15 semanas después de interrumpir el tratamiento.
El embarazo
No hay datos suficientes sobre el uso de ustekinumab durante el embarazo. Los estudios en animales no indican efectos nocivos directos o indirectos con respecto al embarazo, desarrollo embrionario / fetal, parto o desarrollo posnatal (ver sección 5.3). Como medida de precaución, es Es preferible evitar el uso de STELARA durante el embarazo.
Hora de la comida
Se desconoce si ustekinumab se excreta en la leche materna. Algunos estudios clínicos en animales han demostrado la excreción de niveles bajos de ustekinumab en la leche materna. No se sabe si ustekinumab se absorbe sistémicamente después de la ingestión. Dada la capacidad de ustekinumab para desencadenar reacciones adversas en los lactantes, la decisión de interrumpir la lactancia durante el tratamiento y hasta 15 semanas después de la interrupción, o la administración de la terapia con STELARA, debe tomarse teniendo en cuenta el beneficio del tratamiento. " bebé y el beneficio del tratamiento STELARA para la madre.
Fertilidad
No se han evaluado los efectos de ustekinumab sobre la fertilidad humana (ver sección 5.3).
04.7 Efectos sobre la capacidad para conducir y utilizar máquinas.
STELARA tiene una influencia nula o insignificante sobre la capacidad para conducir o utilizar máquinas.
04.8 Efectos indeseables -
Resumen del perfil de seguridad
Las reacciones adversas más frecuentes con ustekinumab (> 5%) en las fases controladas de los ensayos clínicos de psoriasis, artritis psoriásica y enfermedad de Crohn en adultos fueron nasofaringitis y dolor de cabeza. La mayoría se consideraron leves y no fue necesario interrumpir el tratamiento del estudio. Las reacciones adversas que se han notificado con STELARA son reacciones de hipersensibilidad graves, incluida la anafilaxia (ver sección 4.4). El perfil de seguridad general fue similar para los pacientes con psoriasis, artritis psoriásica y enfermedad de Crohn.
Tabla resumen de reacciones adversas
Los datos de seguridad informados a continuación reflejan la exposición a ustekinumab en adultos en 12 estudios clínicos de fase II y fase III en los que participaron 5.884 pacientes (4.135 con psoriasis y / o artritis psoriásica y 1.749 con enfermedad de Crohn). Esto incluye la exposición a STELARA en las fases controladas y no controladas de ensayos clínicos durante al menos 6 meses o 1 año (4.105 y 2.846 pacientes con psoriasis, artritis psoriásica o enfermedad de Crohn respectivamente) con exposición durante al menos 4 o 5 años (1.482 y 838 respectivamente) pacientes con psoriasis).
La Tabla 3 proporciona una lista de reacciones adversas de ensayos clínicos en psoriasis, artritis psoriásica y enfermedad de Crohn en adultos, así como reacciones adversas notificadas en la experiencia post-comercialización. Las reacciones adversas al fármaco se han enumerado por sistema de clasificación de órganos y frecuencia, utilizando la siguiente convención: Muy frecuentes (≥ 1/10), Frecuentes (≥ 1/100 a
Dentro de cada clase de frecuencia, las reacciones adversas se notifican en orden decreciente de gravedad.
Tabla 3 - Lista de reacciones adversas
Descripción de reacciones adversas seleccionadas
Infecciones
En algunos estudios controlados con placebo en pacientes con psoriasis, artritis psoriásica y enfermedad de Crohn, la frecuencia de infección o infección grave fue similar entre los pacientes tratados con ustekinumab y los tratados con placebo. En la fase de tratamiento con placebo en los ensayos clínicos en pacientes con psoriasis, pacientes con artritis psoriásica y pacientes con enfermedad de Crohn, la frecuencia de infección fue de 1,38 por paciente-año de hacer un seguimiento en pacientes que recibieron ustekinumab y 1,35 en los que recibieron placebo. Se produjeron casos de infecciones graves en un grado de 0,03 por paciente-año de hacer un seguimiento en pacientes tratados con ustekinumab (27 infecciones graves en 829 pacientes-año de hacer un seguimiento) y 0,03 en pacientes tratados con placebo (11 infecciones graves en 385 pacientes-año de hacer un seguimiento) (ver sección 4.4).
En las fases controladas y no controladas de los ensayos clínicos en psoriasis, artritis psoriásica y enfermedad de Crohn, que representan 10.953 pacientes-año de exposición en 5.884 pacientes, el hacer un seguimiento la mediana fue de 0,99 años; 3,2 años para los estudios de psoriasis, 1,0 año para los estudios de artritis psoriásica y 0,6 años para los estudios de la enfermedad de Crohn. La frecuencia de infección fue de 0,91 por paciente-año de hacer un seguimiento en pacientes tratados con ustekinumab y la frecuencia de infecciones graves fue de 0,02 por paciente-año de hacer un seguimiento en pacientes que reciben ustekinumab (178 infecciones graves en 10953 pacientes-año de hacer un seguimiento) y las infecciones graves notificadas incluyeron absceso anal, celulitis, neumonía, diverticulitis, gastroenteritis e infecciones virales.
En los ensayos clínicos, los pacientes con tuberculosis latente que fueron tratados concomitantemente con isoniazida no desarrollaron tuberculosis.
Neoplasias
En las fases controladas con placebo de los ensayos clínicos en psoriasis, artritis psoriásica y enfermedad de Crohn, la incidencia de neoplasias, excluyendo el cáncer de piel no melanoma, fue de 0,12 por 100 pacientes-año de hacer un seguimiento para los pacientes tratados con ustekinumab (1 paciente de 829 pacientes-año de hacer un seguimiento) en comparación con 0,26 para los pacientes tratados con placebo (1 paciente de 385 pacientes-año de hacer un seguimiento). La incidencia de cáncer de piel no melanoma fue de 0,48 por 100 pacientes-año hacer un seguimiento para pacientes en terapia con ustekinumab (4 pacientes de 829 pacientes-año de hacer un seguimiento) en comparación con 0,52 para los pacientes tratados con placebo (2 pacientes de 385 pacientes-año de hacer un seguimiento).
En las fases controladas y no controladas de los ensayos clínicos en psoriasis, artritis psoriásica y enfermedad de Crohn, que representan 10.935 pacientes-año de exposición en 5.884 pacientes, el hacer un seguimiento la mediana fue de 1,0 año; 3,2 años para los estudios de psoriasis, 1,0 años para los estudios de artritis psoriásica y 0,6 años para los estudios de enfermedad de Crohn. Se notificaron neoplasias, excluyendo el cáncer de piel no melanoma, en 58 pacientes en 10.935 pacientes-año de hacer un seguimiento (incidencia de 0,53 por 100 pacientes-año de hacer un seguimiento para pacientes tratados con ustekinumab). La incidencia de neoplasias malignas notificadas en pacientes tratados con ustekinumab es comparable a la incidencia esperada en la población general (tasa de incidencia estandarizada = 0,87 [intervalo de confianza del 95%: 0,66, 1,14], corregido por edad, sexo y raza). Las neoplasias malignas observadas con mayor frecuencia, distintas del cáncer de piel no melanoma, fueron el cáncer de próstata, el melanoma, el cáncer colorrectal y el cáncer de mama. La incidencia de cáncer de piel no melanoma fue de 0,49 por 100 pacientes-año de hacer un seguimiento para los pacientes tratados con ustekinumab (53 pacientes de 10919 pacientes-año de hacer un seguimiento). La proporción de pacientes con cánceres de piel de células basales y de células escamosas (4: 1) es comparable con la proporción esperada en la población general (ver sección 4.4).
Reacciones hipersensibles
Durante las fases controladas de los ensayos clínicos de psoriasis y artritis psoriásica de ustekinumab, sarpullido y urticaria se observaron en
Inmunogenicidad
En ensayos clínicos en psoriasis y artritis psoriásica, menos del 8% de los pacientes que tomaban ustekinumab desarrollaron anticuerpos contra ustekinumab. En los ensayos clínicos sobre la enfermedad de Crohn, menos del 3% de los pacientes tratados con ustekinumab desarrollaron anticuerpos contra ustekinumab. No se observó asociación aparente entre el desarrollo de anticuerpos contra ustekinumab y el desarrollo de reacciones en el lugar de la inyección. La mayoría de los pacientes positivos para anticuerpos antiustekinumab tenían anticuerpos neutralizantes. La eficacia del tratamiento tendió a ser menor en pacientes positivos. sin embargo, la positividad de anticuerpos no excluyó una respuesta clínica.
Población pediátrica
Reacciones adversas en pacientes pediátricos a partir de 12 años con psoriasis en placas
La seguridad de ustekinumab se estudió en un estudio de fase 3 en el que participaron 110 pacientes de 12 a 17 años durante un máximo de 60 semanas. Los eventos adversos informados en este estudio fueron similares a los observados en estudios anteriores en adultos con psoriasis en placas.
Notificación de sospechas de reacciones adversas
La notificación de sospechas de reacciones adversas que se produzcan después de la autorización del medicamento es importante, ya que permite un seguimiento continuo de la relación beneficio / riesgo del medicamento. Se ruega a los profesionales sanitarios que notifiquen cualquier sospecha de reacciones adversas a través del sistema nacional de notificación.
04.9 Sobredosis -
En estudios clínicos, se han administrado por vía intravenosa dosis únicas del medicamento de hasta 6 mg / kg, sin observar la aparición de toxicidad limitante de la dosis. En caso de sobredosis, se recomienda que se controle al paciente para detectar cualquier signo o síntoma de reacciones adversas y se instaure inmediatamente la terapia sintomática adecuada.
05.0 PROPIEDADES FARMACOLÓGICAS -
05.1 "Propiedades farmacodinámicas -
Grupo farmacoterapéutico: inmunosupresores, inhibidores de interleucina, código ATC: L04AC05.
Mecanismo de acción
Ustekinumab es un anticuerpo monoclonal IgG1κ completamente humano que se une específicamente a la proteína p40, subunidad compartida de las citocinas humanas interlukin (IL) -12 e IL-23. Ustekinumab inhibe la actividad biológica de IL-12 e IL-23 humanas al evitar la unión de p40 a la proteína receptora IL-12Rb1 expresada en la superficie de las células inmunes. Ustekinumab no puede unirse a IL-12 o IL-23 que ya están unidas a los receptores de IL-12Rb1 presentes en la superficie celular. Por lo tanto, es poco probable que ustekinumab contribuya a la citotoxicidad mediada por el complemento o mediada por anticuerpos de las células con receptores de IL-12 y / o IL-23. IL-12 e IL-23 son heterodiméricos citocinas secretadas por células presentadoras de antígenos activadas, como macrófagos y células dendríticas, y ambas citocinas participan en la actividad inmunitaria; la IL-12 estimula las células asesino natural (NK) y conduce a la diferenciación de las células T CD4 + hacia el fenotipo T ayudante 1 (Th1), IL-23 induce la ruta de T ayudante 17 (Th17). Sin embargo, la regulación anormal de IL-12 e IL-23 se ha asociado con enfermedades inmunomediadas, como psoriasis, artritis psoriásica y enfermedad de Crohn.
Al unirse a la subunidad p40 compartida de IL-12 e IL-23, ustekinumab puede ejercer sus efectos clínicos en la psoriasis, la artritis psoriásica y la enfermedad de Crohn al interrumpir las vías de las citocinas Th1 y Th17, que son cruciales para la enfermedad de estas enfermedades. En pacientes con enfermedad de Crohn, el tratamiento con ustekinumab dio como resultado una disminución de los índices inflamatorios, incluida la proteína C reactiva (PCR) y la calprotectina fecal, durante la fase de inducción; esta inducción se mantuvo luego durante la fase de mantenimiento.
Inmunización
Durante la extensión a largo plazo del estudio de psoriasis 2 (PHOENIX 2), los pacientes adultos tratados con STELARA durante al menos 3,5 años mostraron respuestas de anticuerpos similares tanto a la vacuna antitetánica como al polisacárido neumocócico como grupo de control de pacientes psoriásicos tratados con fármacos no sistémicos. Una proporción similar de pacientes adultos desarrolló niveles protectores de anticuerpos antineumocócicos y antitetánicos y los títulos de anticuerpos fueron similares entre los pacientes tratados con STELARA y los pacientes del grupo de control.
Eficacia clínica y seguridad
Psoriasis en placas (adultos)
Los perfiles de eficacia y seguridad de ustekinumab se evaluaron en 1.996 pacientes en dos ensayos clínicos aleatorizados, doble ciego y controlados con placebo realizados en pacientes con psoriasis en placas de moderada a grave que eran candidatos a fototerapia o terapia sistémica. Además, un ensayo clínico controlado con tratamiento activo, aleatorizado y cegado por el evaluador comparó ustekinumab y etanercept en pacientes con psoriasis en placas de moderada a grave que respondieron de manera inadecuada o que eran intolerantes o que tenían contraindicaciones para ciclosporina, MTX o PUVA.
El estudio de psoriasis 1 (PHOENIX 1) evaluó a 766 pacientes. De estos, el 53% no habían respondido, eran intolerantes o tenían contraindicaciones para otra terapia sistémica. Los pacientes asignados aleatoriamente a ustekinumab fueron tratados con dosis de 45 mg o 90 mg en las semanas 0 y 4 y posteriormente con la misma dosis cada 12 semanas. Pacientes , que fueron aleatorizados al grupo de tratamiento con placebo en las semanas 0 y 4, cambiaron a ustekinumab (45 mg o 90 mg) en las semanas 12 y 16, seguido de una dosis cada 12 semanas. Los pacientes originalmente aleatorizados a ustekinumab, que lograron una respuesta de 75 en el índice Índice de gravedad y área de la psoriasis (PASI) (mejoría en PASI de al menos 75% desde el inicio) en las semanas 28 y 40, fueron reasignados al azar y asignados al grupo de tratamiento con ustekinumab, administrado cada 12 semanas, o al grupo de placebo (es decir, suspensión de la terapia) . Los pacientes reasignados al grupo de placebo en la semana 40 reiniciaron ustekinumab con su esquema de dosificación original si experimentaron una pérdida de al menos el 50% de la mejoría PASI lograda en la semana 40. Todos los pacientes fueron seguidos durante un total de 76 semanas después la primera administración del fármaco del estudio.
El estudio de psoriasis 2 (PHOENIX 2) evaluó a 1230 pacientes. De estos, el 61% no respondía, era intolerante o tenía contraindicaciones para "otra terapia sistémica. Los pacientes asignados al azar a ustekinumab fueron tratados con dosis de 45 mg o 90 mg en las semanas 0 y 4 y luego con una dosis adicional en la semana 16". fueron aleatorizados al grupo de tratamiento con placebo en las semanas 0 y 4 se cambiaron a ustekinumab (45 mg o 90 mg) en las semanas 12 y 16. Todos los pacientes fueron seguidos durante un total de 52 semanas después de la primera administración del tratamiento del estudio.
El estudio de psoriasis 3 (ACCEPT) evaluó a 903 pacientes con psoriasis moderada a grave que respondieron inadecuadamente o que eran intolerantes o que tenían contraindicaciones a otras terapias sistémicas, comparando la eficacia de ustekinumab frente a etanercept y evaluando la seguridad de los dos biológicos en los pacientes. Durante el período de control activo de 12 semanas del estudio, los pacientes fueron aleatorizados para recibir etanercept (50 mg dos veces por semana), ustekinumab 45 mg en las semanas 0 y 4, o ustekinumab 90 mg en las semanas 0 y 4.
En los ensayos clínicos de psoriasis 1 y 2, las características basales de la enfermedad generalmente se superpusieron en todos los grupos de tratamiento con una puntuación PASI basal mediana que variaba de 17 a 18, un "área psoriásica de la superficie corporal (Área superficial del cuerpo, BSA) mediana ≥ 20 y una mediana de la puntuación del índice de calidad de vida dermatológica (Índice de calidad de vida en dermatología, DLQI) entre 10 y 12. Aproximadamente un tercio (estudio de psoriasis 1) y una cuarta parte (estudio de psoriasis 2) de los pacientes tenían artritis psoriásica (APs). También se observó una gravedad de la enfermedad similar en el Estudio 3 de psoriasis.
L "punto final En estos estudios, lo principal fue la proporción de pacientes que alcanzaron una respuesta PASI 75 desde el inicio en la semana 12 (ver Tablas 4 y 5).
Tabla 4 - Resumen de la respuesta clínica en el estudio de psoriasis 1 (PHOENIX 1) y el estudio 2 (PHOENIX 2)
en P
b PGA = (Evaluación global del médico) evaluación global del médico
Tabla 5 - Resumen de la respuesta clínica en la semana 12 en el estudio de psoriasis 3 (ACCEPT)
en P
b p = 0,012 para ustekinumab 45 mg versus etanercept.
En el estudio de psoriasis 1, el mantenimiento de una puntuación PASI 75 fue significativamente mayor con la continuación del tratamiento que con la interrupción del mismo (p
En los pacientes reasignados al azar a placebo que reiniciaron ustekinumab en su régimen original después de una pérdida ≥ 50% de la mejoría PASI, el 85% recuperó una respuesta PASI 75 dentro de las 12 semanas posteriores a la reintroducción del tratamiento. En el estudio de psoriasis 1, en la semana 2 y la semana 12, se observaron mejoras significativas en el DLQI inicial en cada grupo de tratamiento con ustekinumab en comparación con el grupo de placebo. La mejora se mantuvo hasta la semana 28. De manera similar, se observaron mejoras significativas en el estudio de psoriasis 2 en las semanas 4 y 12, que se mantuvieron hasta la semana 24. En el estudio de psoriasis 1, las mejoras en la psoriasis también fueron significativas. Psoriasis ungueal (índice NAPSI, Índice de gravedad de la psoriasis ungueal), las puntuaciones generales del componente mental y físico del SF-36 y la escala analógica visual (Escala analógica visual, EVA) para el prurito en cada grupo de tratamiento con ustekinumab en comparación con placebo. En el estudio de psoriasis 2, la escala HADS (Escala hospitalaria de ansiedad y depresión) y el cuestionario WLQ (Cuestionario de limitaciones laborales) en cada grupo de tratamiento con ustekinumab versus placebo.
Artritis psoriásica (PsA) (adultos)
Se ha demostrado que ustekinumab mejora los signos y síntomas, la función física y la calidad de vida relacionada con la salud y reduce la tasa de progresión del daño articular periférico en pacientes adultos con APs activa.
Se evaluó la seguridad y eficacia de ustekinumab en 927 pacientes en dos ensayos clínicos aleatorizados, doble ciego y controlados con placebo en pacientes con APs activa (≥ 5 articulaciones inflamadas y ≥ 5 dolorosas) a pesar del tratamiento antiinflamatorio no esteroideo (AINE). ) o tratamiento con fármacos antirreumáticos modificadores de la enfermedad (FAME). Los pacientes de estos estudios habían sido diagnosticados con APs durante al menos 6 meses. Se inscribieron pacientes con cualquier subtipo de APs, incluida la artritis poliarticular sin evidencia de nódulos remautoides (39%), espondilitis con artritis periférica (28%), artritis asimétrica periférica (21%), afectación de las articulaciones interfalángicas distales (12%) y artritis mutilante (0,5%). Más del 70% y el 40% de los pacientes en ambos estudios tenían entesitis y dactilitis en línea de base, respectivamente. Los pacientes fueron aleatorizados para recibir ustekinumab 45 mg, 90 mg o placebo por vía subcutánea en las semanas 0 y 4 seguidos de un
administración cada 12 semanas (q12w). Aproximadamente el 50% de los pacientes continuaron con dosis estables de MTX (≤ 25 mg / semana).
En el Estudio 1 de PsA (PSUMMIT I) y el Estudio 2 de PsA (PSUMMIT II), el 80% y el 86% de los pacientes, respectivamente, habían sido tratados previamente con FAME. En el Estudio 1 no se permitió el tratamiento previo con agentes antifactor de necrosis tumoral (TNF) α. En el Estudio 2, la mayoría de los pacientes (58%, n = 180) habían recibido previamente uno o más tratamientos con un agente anti-TNFα, de los cuales más del 70% había interrumpido el tratamiento anti-TNFα en cualquier momento por pérdida de eficacia o intolerancia.
Signos y síntomas
El tratamiento con ustekinumab produjo mejoras significativas en la evaluación de la actividad de la enfermedad en comparación con el placebo en la semana 24. El criterio de valoración principal fue el porcentaje de pacientes que alcanzaron la respuesta del American College of Rheumatology (ACR) 20 en la semana 24. I Los resultados clave de eficacia se muestran en la siguiente Tabla 6 . Tabla 6 - Número de pacientes que lograron una respuesta clínica en el Estudio 1 de artritis psoriásica (PSUMMIT I) y el Estudio 2 (PSUMMIT II) en la semana 24
en P
b p
c p = NS
d Número de pacientes con afectación de psoriasis cutánea basal BSA ≥ 3%
Las respuestas ACR 20, 50 y 70 mejoraron continuamente o se mantuvieron constantes durante la semana 52 (estudio 1 y 2 de PsA) y la semana 100 (estudio 1 de PsA). En el estudio 1 de PsA, las respuestas ACR 20 en la semana 100 se lograron en un 57% y un 64%, para 45 mg y 90 mg, respectivamente. En el estudio 2 de PsA, las respuestas ACR 20 en la semana 52 se lograron en un 47% y un 48%, para 45 mg y 90 mg, respectivamente.
El porcentaje de pacientes que lograron una respuesta según los Criterios de respuesta de artritis psoriásica modificada (PsARC) también fue significativamente mayor en el grupo de ustekinumab en comparación con el placebo en la semana 24. Las respuestas de PsARC se mantuvieron hasta las semanas 52 y 100. Un porcentaje "alto" de ustekinumab- Los pacientes tratados que tenían espondilitis con artritis periférica como presentación primaria, mostraron una mejora del 50 y 70 por ciento en la puntuación del Índice de Actividad de la Enfermedad de la Espondilitis Anquilosante de Bath (BASDAI) en comparación con el placebo en la semana 24. El tratamiento con ustekinumab fue similar entre los pacientes que recibieron MTX concomitante y los que no recibieron MTX y se mantuvieron hasta las semanas 52 y 100. Los pacientes tratados previamente con agentes anti-TNFα que recibieron ustekinumab lograron una mayor respuesta en la semana 24 en comparación con los pacientes que recibieron el placebob o (la respuesta ACR 20 en la semana 24 para 45 mg y 90 mg fue 37% y 34%, respectivamente, en comparación con placebo 15%; pag
Para los pacientes con entesitis y / o dactilitis al inicio del estudio, se observó una mejora significativa en la puntuación de entesitis y dactilitis en el grupo de ustekinumab en comparación con el grupo de placebo en la semana 24 en el estudio PsA 2. Mejora significativa en la puntuación de entesitis y numérica (no estadísticamente significativa ) mejora en la puntuación de dactilitis en el grupo de ustekinumab 90 mg (p = NS) en comparación con placebo en la semana 24. Las mejoras en la puntuación de entesitis y dactilitis se mantuvieron hasta las semanas 52 y 100.
Respuesta radiográfica
El daño estructural en ambas manos y pies se expresó como el cambio en la puntuación total de van der Heijde-Sharp (puntuación vdH-S), modificada para PsA al agregar las articulaciones interfalángicas distales de la mano, desde la línea de base. Se realizó un análisis integrado preespecífico combinando datos de 927 sujetos tanto del Estudio 1 de PsA como del Estudio 2.
Ustekinumab demostró una disminución estadísticamente significativa en la tasa de progresión del daño estructural en comparación con el placebo, medida por el cambio desde el inicio hasta la semana 24 en la puntuación total modificada de vdH-S (la puntuación media ± DE fue 0,97 ± 3,85 en el grupo de placebo frente a 0,40 ± 2,11 y 0,39 ± 2,40 en los grupos de ustekinumab 45 mg (p
Función física y calidad de vida relacionada con la salud
Los pacientes tratados con ustekinumab mostraron una mejora significativa en la función física según la evaluación del Índice de discapacidad del Cuestionario de evaluación de la salud (HAQ-DI) en la semana 24. También el porcentaje de pacientes que lograron una mejoría clínicamente significativa ≥ 0,3 en la puntuación HAQ-DI desde el inicio fue significativamente mayor en el grupo de ustekinumab que en el grupo de placebo La mejora en la puntuación HAQ-DI desde el inicio se mantuvo hasta las semanas 52 y 100.
C "fue una mejora significativa en la puntuación DLQI en el grupo de ustekinumab en comparación con el placebo en la semana 24, que se mantuvo durante las semanas 52 y 100. En el estudio PsA 2 c" fue una mejora significativa en la puntuación de la evaluación funcional crónica. Terapia de la enfermedad - Fatiga (FACIT-F) en el grupo de ustekinumab en comparación con el grupo de placebo en la semana 24. El porcentaje de pacientes que lograron una mejora significativa en la fatiga (4 puntos en FACIT-F) también fue significativamente mayor en el grupo de ustekinumab en comparación con el placebo. Las mejoras en la puntuación FACIT se mantuvieron hasta la semana 52.
Población pediátrica
La Agencia Europea de Medicamentos ha aplazado la obligación de presentar los resultados de los estudios con ustekinumab en uno o más subconjuntos de la población pediátrica de 6 a 11 años para la psoriasis en placas de moderada a grave y la artritis idiopática juvenil (ver sección 4.2 para obtener información sobre el uso pediátrico). .
Psoriasis en placas en pacientes pediátricos
Se ha demostrado que ustekinumab mejora los signos y síntomas relacionados con la salud y la calidad de vida en pacientes pediátricos de 12 años o más con psoriasis en placas.
La eficacia de ustekinumab se estudió en 110 pacientes pediátricos de 12 a 17 años con psoriasis en placas de moderada a grave en un estudio de fase 3, multicéntrico, aleatorizado, doble ciego, controlado con placebo (CADMUS). Los pacientes fueron aleatorizados para tomar placebo (n = 37), ya sea la dosis recomendada de ustekinumab (ver sección 4.2; n = 36) o la mitad de la dosis recomendada de ustekinumab (n = 37) por inyección subcutánea en las semanas 0 y 4 y posteriormente cada 12 semanas (q12w) En la semana 12, placebo Los pacientes tratados fueron cambiados a tratamiento con ustekinumab.
Los pacientes con PASI ≥ 12, PGA ≥ 3 y afectación de BSA de al menos el 10% que eran candidatos para terapia sistémica o fototerapia fueron elegibles para el estudio. Aproximadamente el 60% de los pacientes tuvo exposición previa a la terapia sistémica convencional o fototerapia y aproximadamente el 11% de los pacientes tuvo exposición previa a productos biológicos.
El criterio de valoración principal fue la proporción de pacientes que alcanzaron un índice de PGA en la semana 12 despejado o mínimo . Los criterios de valoración secundarios incluyeron PASI 75, PASI 90, cambio desde el inicio en Índice de calidad de vida en dermatología infantil (CDLQI), cambio con respecto al valor inicial en la puntuación total de PedsQL (Inventario de calidad de vida pediátrica) en la Semana 12. En la Semana 12, los sujetos tratados con ustekinumab mostraron una mejoría significativamente mayor en su psoriasis y calidad de vida relacionada con la salud que los sujetos tratados con placebo (Tabla 7).
Se realizó un seguimiento de la eficacia de todos los pacientes hasta 52 semanas después de la primera administración del agente del estudio. El porcentaje de pacientes con una puntuación PGA despejado o mínimo y la proporción de pacientes que alcanzaron PASI 75 mostró una brecha entre los grupos de ustekinumab y placebo en la primera visita post-basal en la Semana 4, alcanzando su punto máximo en la Semana 12. Las mejoras en PGA, PASI, CDLQI y PedsQL se mantuvieron en la Semana 52 ( Tabla 7).
Tabla 7: Resumen de los criterios de valoración primarios y secundarios en la semana 12 y la semana 52
en P
b CDLQI: CDLQI es una herramienta dermatológica para evaluar el efecto de un problema de la piel en la calidad de vida relacionada con la salud en la población pediátrica. CDLQI de 0 o 1 indica que no hay efecto sobre la calidad de vida del niño.
c p = 0,002
d PedsQL: PedsQL es una medida general de la calidad de vida relacionada con la salud desarrollada para su uso en niños y adolescentes.
y p = 0,028
Durante el período controlado con placebo hasta la semana 12, la eficacia en ambos grupos a la dosis recomendada y la mitad de la dosis recomendada fue generalmente comparable con respecto al criterio de valoración principal (69,4% y 67,6%, respectivamente), aunque hubo evidencia de una dosis -Respuesta relacionada con criterios de eficacia de nivel superior (por ejemplo, PGA despejado , PASI 90). Después de la semana 12, la eficacia fue generalmente mayor y mejor sostenida en el grupo de tratamiento que recibió la dosis completa recomendada que en el grupo que recibió la mitad, en el que la pérdida moderada de eficacia observada al final del tratamiento fue más frecuente, cada intervalo de dosis de 12 semanas. El perfil de seguridad de la dosis recomendada y la mitad de la dosis recomendada fue comparable.
enfermedad de Crohn
La seguridad y eficacia de ustekinumab se evaluaron en tres estudios multicéntricos, aleatorizados, doble ciego y controlados con placebo en pacientes adultos con enfermedad de Crohn activa de moderada a grave (índice de actividad de la enfermedad de Crohn [CDAI] = índice de actividad de la enfermedad de Crohn ≥ 220 y ≤ 450 ). El programa de desarrollo clínico consistió en dos estudios de inducción intravenosa de 8 semanas (UNITED-1 y UNITED-2) seguidos de un estudio de mantenimiento subcutáneo aleatorizado de 44 semanas (IM-UNITED) que constaba de 52 semanas de tratamiento. Los estudios de inducción involucraron a 1.409 pacientes (UNITED-1, n = 769; UNITED-2 n = 640). El criterio de valoración principal de ambos estudios de inducción fue la proporción de sujetos en respuesta clínica (definida como una reducción del CDAI en ≥ 100 puntos) en la semana 6. Se recopilaron y analizaron datos de eficacia hasta la semana 8 para ambos estudios. Se permitieron dosis concomitantes de corticosteroides orales, inmunomoduladores, aminosalicilatos y antibióticos y el 75% de los pacientes continuó recibiendo al menos uno de estos fármacos. En ambos estudios, los pacientes fueron aleatorizados para recibir una dosis intravenosa única de una dosis recomendada de aproximadamente 6 mg / kg (ver sección 4.2 de la ficha técnica de STELARA 130 mg concentrado para solución para perfusión), o una dosis fija de 130 mg / kg. mg de ustekinumab o placebo en la semana 0.
Los pacientes tratados con UNITED-1 no respondieron o fueron intolerantes a la terapia anti-TNFα previa. Aproximadamente el 48% de los pacientes no respondió a la terapia previa con un anti-TNF-α y el 52% no respondió a las terapias previas con 2 o 3 anti-TNF-α. En este estudio, el 29,1% de los pacientes tuvo una respuesta inicial inadecuada (no respondedores primarios), el 69,4% respondió pero "perdió la respuesta" (no respondedores secundarios) y el 36,4% no toleraba las terapias anti-TNFa.
Los pacientes tratados con UNITED-2 no han respondido al menos a una terapia convencional, incluidos corticosteroides o inmunomoduladores, y no habían recibido tratamiento previo con anti-TNF-α (68,6%) o habían recibido previamente, pero no habían fallado, una terapia anti-TNFα (31,4%).
Tanto en UNITED-1 como en UNITED-2, una proporción significativamente mayor de pacientes se encontraba en respuesta clínica y en remisión en el grupo de ustekinumab en comparación con el placebo (Tabla 8). Las respuestas clínicas y las remisiones fueron significativas desde la semana 3 en los pacientes tratados con ustekinumab y continuaron mejorando hasta la semana 8. En estos estudios de inducción, la eficacia fue mayor y se mantuvo mejor en el grupo de dosis variable que en el grupo con la dosis de 130 mg y por tanto, se recomienda una dosis variable para la inducción intravenosa.
Tabla 8: Inducción de respuesta clínica y remisión en UNITED-1 y UNITED-2
La remisión clínica se define como el índice CDAI
Respuesta 70 puntos se define como una reducción del índice CDAI en al menos 70 puntos
* Fallos anti-TNFα
** fallas de la terapia convencional
en P
b p
El estudio de mantenimiento (IM-UNITED) evaluó a 388 pacientes que lograron una respuesta clínica de 100 puntos en la semana 8 de inducción de ustekinumab en los estudios UNITED-1 y UNITED-2. Los pacientes fueron aleatorizados a un régimen de mantenimiento subcutáneo de 90 mg de ustekinumab cada 8 semanas o 90 mg de ustekinumab cada 12 semanas o placebo durante 44 semanas (para la dosis de mantenimiento recomendada, ver sección 4.2). Un mayor porcentaje de pacientes mantuvo la remisión clínica y la respuesta clínica en los grupos de ustekinumab en comparación con el grupo de placebo en la semana 44 (ver Tabla 9).
Tabla 9: Mantenimiento de la respuesta clínica y la remisión en IM-Uniti (semana 44; 52 semanas desde el inicio de la dosis de inducción)
La remisión clínica se define como el índice CDAI
* El grupo de placebo consistió en pacientes que respondieron a ustekinumab y fueron aleatorizados para recibir placebo al comienzo de la terapia de mantenimiento.
† Pacientes que tenían una respuesta clínica de 100 puntos de ustekinumab al inicio de la terapia de mantenimiento
‡ Pacientes que han fracasado con la terapia convencional pero no con la terapia anti-TNF α
§ Pacientes refractarios / intolerantes a anti-TNF α
en P
b p
c nominalmente significativo (p
En el IM-UNITED, 29 de 129 pacientes no mantuvieron la respuesta a ustekinumab cuando fueron tratados cada 12 semanas y se les permitió ajustar la dosis para recibir ustekinumab cada 8 semanas.
La pérdida de respuesta se definió como un CDAI ≥ 220 puntos y un aumento ≥ 100 puntos en el CDAI desde el inicio. En estos pacientes, se logró la remisión clínica en el 41,4% de los pacientes 16 semanas después del tratamiento.
Los pacientes que no tuvieron respuesta clínica después de la inducción con ustekinumab en la semana 8 en los estudios de inducción UNITED-1 y UNITED-2 (476 pacientes) ingresaron a la parte no aleatorizada del estudio de mantenimiento (IM-UNITED) y luego recibieron una inyección subcutánea de 90 mg de ustekinumab. Ocho semanas después, el 50,5% de los pacientes lograron una respuesta clínica y continuaron recibiendo la dosis de mantenimiento cada 8 semanas; entre estos pacientes que recibieron dosis de mantenimiento continuada, la mayoría mantuvo la respuesta (68,1%) y logró la remisión (50,2%) en la semana 44, en tasas similares a las de los pacientes que respondieron inicialmente a la inducción con ustekinumab.
De los 131 pacientes que respondieron a ustekinumab en la fase de inducción y que fueron aleatorizados al grupo de placebo al comienzo del estudio de mantenimiento, 51 posteriormente no respondieron y recibieron ustekinumab 90 mg por vía subcutánea cada 8 semanas. respuesta y reiniciar ustekinumab lo hizo dentro de las 24 semanas posteriores a la infusión de inducción. De estos 51 pacientes, el 70,6% logró una respuesta clínica y el 39,2% logró la remisión clínica 16 semanas después de recibir la primera dosis subcutánea de ustekinumab.
Endoscopia
El aspecto endoscópico de la mucosa se evaluó en un subestudio en 252 pacientes elegibles con actividad endoscópica inicial de la enfermedad. El criterio principal de valoración fue el cambio desde el valor inicial en la escala simplificada de gravedad de la enfermedad endoscópica para la enfermedad de Crohn (SES-CD), un índice compuesto de los 5 segmentos ileo-cólicos de presencia / tamaño de úlceras, porcentaje de superficie mucosa cubierta por úlceras, porcentaje de superficie mucosa afectada por cualquier otra lesión y presencia / tipo de estrechamiento / estenosis. En la semana 8, después de una única dosis de inducción intravenosa, el cambio en el índice SES-CD fue mayor en el grupo de ustekinumab (n = 155, cambio medio = -2,8) que en el grupo de placebo (n = 97, cambio medio = -0,7 , p = 0,012).
Respuesta en enfermedad fistulizante
En un subconjunto de pacientes con fístulas drenantes al inicio del estudio (8,8%; n = 26), 12/15 (80%) de los pacientes tratados con ustekinumab lograron una respuesta después de 44 semanas (definida como una reducción ≥ 50% desde el inicio en el estudio de inducción en el número de fístulas drenantes) en comparación con 5/11 (45,5%) expuestas al placebo.
Calidad de vida relacionada con la salud
La calidad de vida relacionada con la salud se evaluó mediante los cuestionarios IBDQ y SF-36. En la semana 8, los pacientes tratados con ustekinumab mostraron mayores mejoras clínicas estadísticamente significativas en el índice total del IBDQ y la puntuación resumida del componente mental SF-36 en UNITED-1 y UNITED-2, y la puntuación resumida del componente físico SF-36 en UNITED -2, en comparación con placebo Estas mejoras se mantuvieron generalmente mejor en los pacientes tratados con ustekinumab en el estudio IM-Uniti hasta la semana 44 en comparación con el placebo.
Población pediátrica
La Agencia Europea de Medicamentos ha aplazado la obligación de presentar los resultados de los estudios con ustekinumab en uno o más subconjuntos de la población pediátrica con enfermedad de Crohn (ver sección 4.2 para obtener información sobre uso pediátrico).
05.2 "Propiedades farmacocinéticas -
Absorción
En sujetos sanos, el tiempo medio para alcanzar la concentración sérica máxima (Tmax) fue de 8,5 días después de una única administración subcutánea de 90 mg. Los valores medios de T de ustekinumab tras una única administración subcutánea de 45 mg o 90 mg en pacientes con psoriasis son comparables a los observados en sujetos sanos.
La biodisponibilidad absoluta de ustekinumab en pacientes con psoriasis después de una única administración subcutánea se estimó en 57,2%.
Distribución
La mediana del volumen de distribución durante la fase terminal (Vz) después de una única administración intravenosa en pacientes con psoriasis osciló entre 57 y 83 ml / kg.
Biotransformación
Se desconoce el proceso metabólico exacto de ustekinumab.
Eliminación
Allí autorización La mediana sistémica (CL) en pacientes con psoriasis después de una única administración intravenosa osciló entre 1,99 y 2,34 ml /morir/ kg.
La vida media media (t1 / 2) de ustekinumab fue de aproximadamente 3 semanas en pacientes con psoriasis, artritis psoriásica o enfermedad de Crohn, y varió de 15 a 32 días en todos los estudios de psoriasis y artritis psoriásica.
En un "análisis del perfil farmacocinético poblacional en pacientes con psoriasis, el autorización aparente (CL / F) y aparente volumen de distribución (V / F) fueron 0,465 L / día y 15,7 L, respectivamente. El CL / F de ustekinumab no se vio afectado por el sexo. El análisis farmacocinético de la población mostró una tendencia hacia el aumento del aclaramiento de ustekinumab en pacientes positivos para anticuerpos anti-ustekinumab.
Linealidad de la dosis
La exposición sistémica de ustekinumab (Cmax y AUC) aumentó bastante de manera proporcional a la dosis después de una única administración intravenosa de dosis que van desde 0,09 mg / kg a 4,5 mg / kg o después de una única administración subcutánea en dosis que van desde aproximadamente 24 mg a 240 mg en pacientes. con psoriasis.
Dosis única versus dosis múltiples
Los perfiles de concentración sérica-tiempo de ustekinumab fueron ampliamente predecibles después de dosis subcutáneas únicas o múltiples. En pacientes con psoriasis, las concentraciones séricas en estado estacionario (estado estable) de ustekinumab se lograron a partir de la semana 28 después de la administración subcutánea en las semanas 0 y 4, seguida de la administración cada 12 semanas. La concentración mínima mediana en estado estacionario (estado estable) estaba entre 0,21 mcg / ml y 0,26 mcg / ml (45 mg) y entre 0,47 mcg / ml y 0,49 mcg / ml (90 mg).
Después de la administración subcutánea cada 12 semanas, no se observó una acumulación aparente de la concentración sérica de ustekinumab con el tiempo. En pacientes con enfermedad de Crohn, después de una dosis intravenosa de ~ 6 mg / kg, se administró una dosis de mantenimiento de 90 mg de ustekinumab por vía subcutánea cada 8 o 12 semanas a partir de la semana 8. La concentración en estado estacionario (estado estable) de ustekinumab se alcanzó al comienzo de la segunda dosis de mantenimiento. La concentración mínima media en estado estacionario (estado estable) de ustekinumab osciló entre 1,97 mg / ml y 2,24 mg / ml y entre 0,61 mg / ml y 0,76 mg / ml para 90 mg de ustekinumab cada 8 semanas o cada 12 semanas, respectivamente. Niveles mínimos de ustekinumab en estado estacionario (estado estable) los resultados de ustekinumab 90 mg cada 8 semanas se asociaron con tasas de remisión clínica más altas que los niveles mínimos en el estado estacionario de 90 mg cada 12 semanas.
Impacto del peso en el perfil farmacocinético
En un "análisis farmacocinético de la población de pacientes utilizando datos de pacientes con psoriasis, se encontró que el peso corporal era la covariable que influía de manera más significativa autorización por ustekinumab. La mediana de CL / F de los pacientes que pesan> 100 kg fue aproximadamente un 55% más alta que la de los pacientes que pesan ≤ 100 kg. La mediana de V / F de los pacientes que pesaban> 100 kg fue aproximadamente un 37% más alta que la de los pacientes que pesaban ≤ 100 kg. Las concentraciones séricas medias más bajas de ustekinumab en los pacientes de mayor peso (> 100 kg) en el grupo de dosis de 90 mg fueron comparables a las de los pacientes de menor peso (≤ 100 kg) en el grupo tratado con la dosis de 45 mg. Se obtuvieron resultados similares a partir de un análisis farmacocinético poblacional confirmatorio utilizando datos de pacientes con artritis psoriásica.
Poblaciones especiales
No se dispone de datos farmacocinéticos en pacientes con disfunción renal o hepática.
No se han realizado estudios clínicos específicos en pacientes de edad avanzada.
El perfil farmacocinético de ustekinumab fue generalmente comparable entre pacientes asiáticos y no asiáticos con psoriasis.
En pacientes con enfermedad de Crohn, la variabilidad del CL de ustekinumab se vio afectada por el peso corporal, el nivel de albúmina sérica, la PCR, el fracaso previo del antagonista del TNF, el sexo, la raza (asiática o no asiática) y la presencia de anticuerpos contra ustekinumab, mientras que el peso corporal fue el principal covariable que afecta el volumen de distribución El uso concomitante de inmunomoduladores no tuvo un impacto significativo en la disposición de ustekinumab. El impacto de estas covariables estadísticamente significativas en sus respectivos parámetros farmacocinéticos estuvo dentro de ± 20% cuando se evaluó en un rango de datos representativo de covariables o categorías que se encuentra dentro de la variabilidad general observada en la PK de ustekinumab. En el análisis farmacocinético de la población de pacientes, no se observaron indicios de un efecto del tabaco o el alcohol sobre el perfil farmacocinético de ustekinumab.
Las concentraciones séricas de ustekinumab en pacientes pediátricos de 12 a 17 años con psoriasis tratados con la dosis recomendada basada en el peso corporal fueron generalmente comparables a las de la población adulta con psoriasis tratada con la dosis recomendada para adultos, mientras que las concentraciones séricas de ustekinumab en pacientes pediátricos los pacientes con psoriasis tratados con la mitad de la dosis recomendada en función del peso corporal fueron generalmente más bajos que en los adultos.
Regulación de las enzimas CYP450
Los efectos de IL-12 o IL-23 sobre la regulación de las enzimas CYP450 se evaluaron en un estudio. in vitro utilizando hepatocitos humanos, este estudio demostró que IL-12 y / o IL-23 a niveles de 10 ng / ml no alteran la actividad enzimática del CYP450 humano (CYP1A2, 2B6, 2C9, 2C19, 2D6 o 3A4; ver sección 4.5 ).
05.3 Datos preclínicos sobre seguridad -
Los datos de los estudios no clínicos no muestran riesgos especiales para los seres humanos (p. Ej., Toxicidad orgánica) según los estudios de toxicidad a dosis repetidas y de toxicidad para el desarrollo y la reproducción, incluidas las evaluaciones de farmacología de seguridad. En estudios de toxicidad para la reproducción y el desarrollo realizados en monos cynomolgus, no se observaron efectos adversos sobre los índices de fertilidad masculina, defectos de nacimiento o toxicidad del desarrollo. No se observaron efectos adversos sobre los índices de fertilidad femenina con el uso de un anticuerpo análogo a IL-12/23 en ratones.
Los niveles de dosis en los estudios con animales fueron hasta aproximadamente 45 veces más altos que la dosis equivalente más alta destinada a ser administrada a pacientes con psoriasis. En los monos, estos niveles se tradujeron en concentraciones séricas máximas que fueron 100 veces o más superiores a las observadas en humanos.
No se han realizado estudios de carcinogenicidad de ustekinumab debido a la ausencia de modelos de anticuerpos apropiados libres de reacción cruzada de IL-12/23 p40 en roedores.
06.0 INFORMACIÓN FARMACÉUTICA -
06.1 Excipientes -
L-histidina
Monohidrocloruro de L-histidina monohidrato
Polisorbato 80
Sacarosa
Agua para preparaciones inyectables
06.2 Incompatibilidad "-
En ausencia de estudios de compatibilidad, este medicamento no debe mezclarse con otros medicamentos.
06.3 Período de validez "-
2 años
06.4 Precauciones especiales de conservación
Conservar en nevera (entre 2 ° C y 8 ° C). No congelar.
Mantenga el vial o la jeringa precargada en el embalaje exterior para proteger el medicamento de la luz.
06.5 Naturaleza del envase primario y contenido del envase.
STELARA 45 mg solución inyectable
0,5 ml de solución en un vial de 2 ml de vidrio tipo I, cerrado con un tapón de goma de butilo.
STELARA 90 mg solución inyectable
1 ml de solución en un vial de 2 ml de vidrio tipo I, cerrado con un tapón de goma de butilo.
STELARA 45 mg solución inyectable en jeringa precargada
0.5 mL de solución en una jeringa de vidrio tipo I de 1 mL, con una aguja de acero no removible protegida por un tapón que contiene caucho natural seco (un derivado del látex). La jeringa está equipada con un dispositivo de seguridad pasivo.
STELARA 90 mg solución inyectable en jeringa precargada
1 mL de solución en una jeringa de vidrio tipo I de 1 mL con una aguja de acero no removible protegida por una tapa que contiene caucho natural seco (un derivado del látex). La jeringa está equipada con un dispositivo de seguridad pasivo.
STELARA está disponible en envases de 1 vial o 1 jeringa precargada.
06.6 Instrucciones de uso y manipulación -
La solución contenida en el vial de STELARA o en la jeringa precargada no debe agitarse. La solución debe inspeccionarse visualmente en busca de partículas o decoloración antes de la administración subcutánea. La solución es de transparente a ligeramente opalescente, de incolora a amarillo pálido y puede contener algunas pequeñas partículas de proteína translúcidas o blancas. No es inusual para las soluciones de proteína. No se debe utilizar el producto si la solución está descolorida u opaca, o si hay partículas extrañas. Antes de la administración, se debe dejar que STELARA alcance la temperatura ambiente (aproximadamente media hora). Las instrucciones de uso detalladas se proporcionan en el prospecto.
STELARA no contiene conservantes, por lo que no debe utilizarse ningún medicamento no utilizado que quede en el vial y la jeringa. STELARA se presenta como un vial estéril de un solo uso o una jeringa precargada de un solo uso. La jeringa, la aguja y el vial nunca deben reutilizarse. El medicamento no utilizado y los desechos de este medicamento deben eliminarse de acuerdo con los requisitos locales.
07.0 TITULAR DE LA "AUTORIZACIÓN DE COMERCIALIZACIÓN" -
Janssen-Cilag International NV
Turnhoutseweg 30
2340 Beerse
Bélgica
08.0 NÚMERO DE AUTORIZACIÓN DE COMERCIALIZACIÓN -
STELARA 45 mg solución inyectable
EU / 1/08/494/001
STELARA 90 mg solución inyectable
EU / 1/08/494/002
STELARA 45 mg solución inyectable en jeringa precargada
EU / 1/08/494/003
STELARA 90 mg solución inyectable en jeringa precargada
EU / 1/08/494/004
038936035
038936047
038936011
038936023
09.0 FECHA DE LA PRIMERA AUTORIZACIÓN O RENOVACIÓN DE LA AUTORIZACIÓN -
Fecha de la primera autorización: 16 de enero de 2009
Fecha de la última renovación: 19 de septiembre de 2013